

An improved relaxed complex scheme for receptor flexibility in computer-aided drug design
- PMID: 18196463
- PMCID: PMC2516539
- DOI: 10.1007/s10822-007-9159-2
An improved relaxed complex scheme for receptor flexibility in computer-aided drug design
Abstract
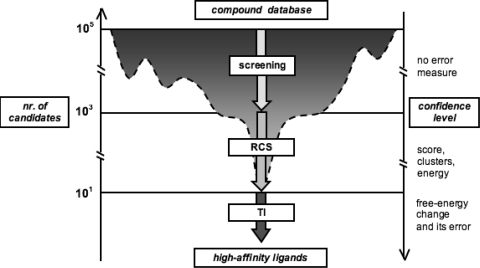
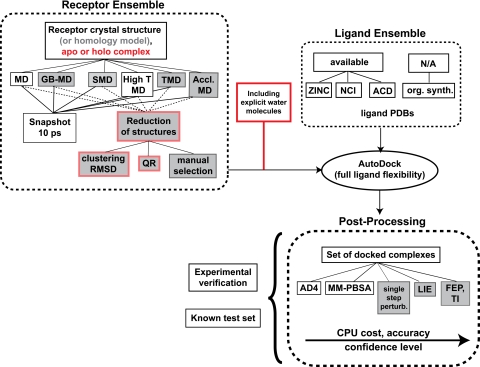
The interactions among associating (macro)molecules are dynamic, which adds to the complexity of molecular recognition. While ligand flexibility is well accounted for in computational drug design, the effective inclusion of receptor flexibility remains an important challenge. The relaxed complex scheme (RCS) is a promising computational methodology that combines the advantages of docking algorithms with dynamic structural information provided by molecular dynamics (MD) simulations, therefore explicitly accounting for the flexibility of both the receptor and the docked ligands. Here, we briefly review the RCS and discuss new extensions and improvements of this methodology in the context of ligand binding to two example targets: kinetoplastid RNA editing ligase 1 and the W191G cavity mutant of cytochrome c peroxidase. The RCS improvements include its extension to virtual screening, more rigorous characterization of local and global binding effects, and methods to improve its computational efficiency by reducing the receptor ensemble to a representative set of configurations. The choice of receptor ensemble, its influence on the predictive power of RCS, and the current limitations for an accurate treatment of the solvent contributions are also briefly discussed. Finally, we outline potential methodological improvements that we anticipate will assist future development.
Figures



References
-
- May A, Zacharias M (2005) Biochim Biophys Acta 1754:225 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
