

PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition
- PMID: 18199835
- PMCID: PMC2242705
- DOI: 10.1073/pnas.0711281105
PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition
Abstract
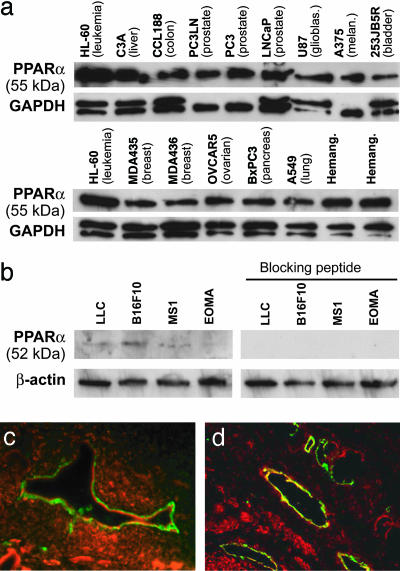
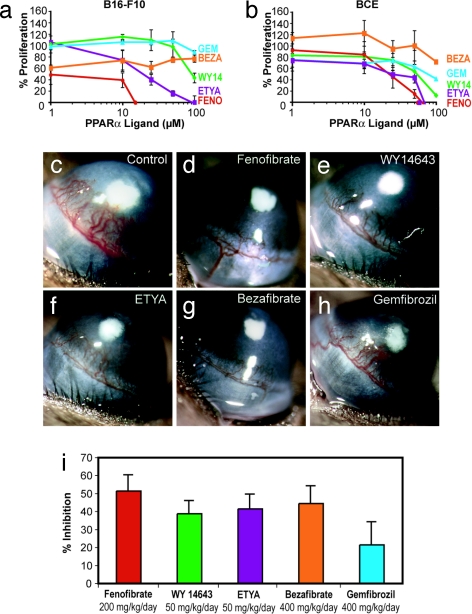
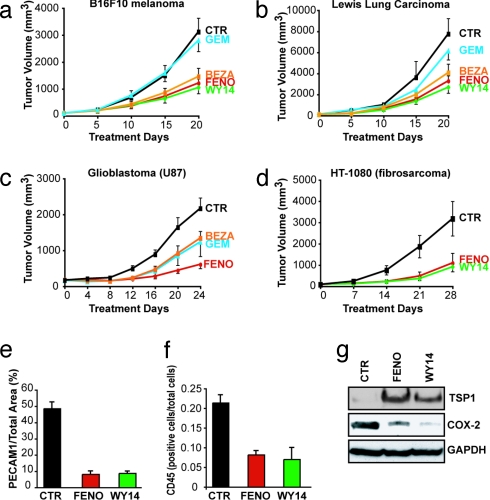
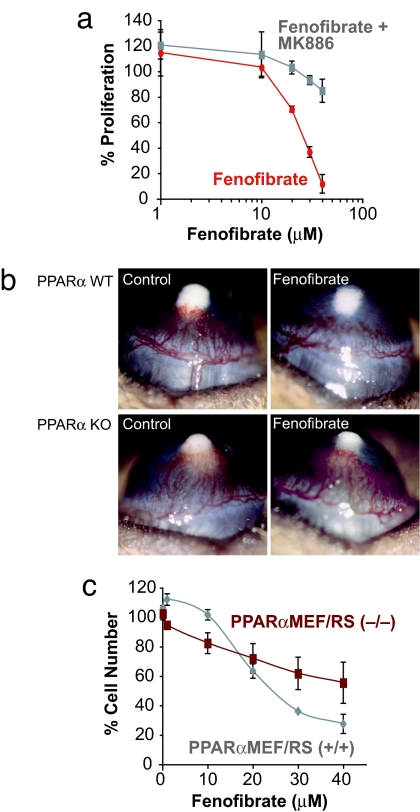
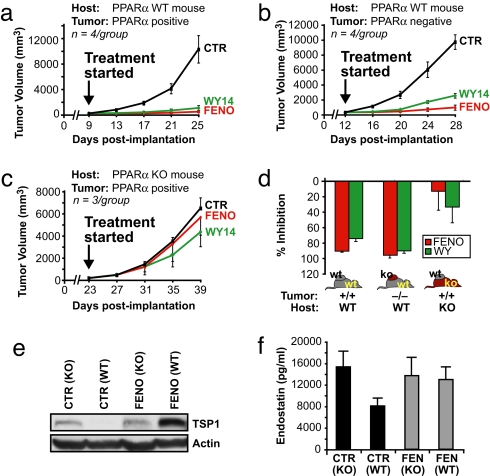
Angiogenesis and inflammation are central processes through which the tumor microenvironment influences tumor growth. We have demonstrated recently that peroxisome proliferator-activated receptor (PPAR)alpha deficiency in the host leads to overt inflammation that suppresses angiogenesis via excess production of thrombospondin (TSP)-1 and prevents tumor growth. Hence, we speculated that pharmacologic activation of PPARalpha would promote tumor growth. Surprisingly, the PPARalpha agonist fenofibrate potently suppressed primary tumor growth in mice. This effect was not mediated by cancer-cell-autonomous antiproliferative mechanisms but by the inhibition of angiogenesis and inflammation in the host tissue. Although PPARalpha-deficient tumors were still susceptible to fenofibrate, absence of PPARalpha in the host animal abrogated the potent antitumor effect of fenofibrate. In addition, fenofibrate suppressed endothelial cell proliferation and VEGF production, increased TSP-1 and endostatin, and inhibited corneal neovascularization. Thus, both genetic abrogation of PPARalpha as well as its activation by ligands cause tumor suppression via overlapping antiangiogenic pathways. These findings reveal the potential utility of the well tolerated PPARalpha agonists beyond their use as lipid-lowering drugs in anticancer therapy. Our results provide a mechanistic rationale for evaluating the clinical benefits of PPARalpha agonists in cancer treatment, alone and in combination with other therapies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Devchand PR, et al. The PPARα-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. - PubMed
-
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPARα in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy 14,643. Carcinogenesis. 1997;18:2029–2033. - PubMed
-
- Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. - PubMed
-
- Collett GP, et al. Peroxisome proliferator-activated receptorα is an androgen-responsive gene in human prostate and is highly expressed in prostatic adenocarcinoma. Clin Cancer Res. 2000;6:3241–3248. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
