

The contribution of c-Jun N-terminal kinase activation and subsequent Bcl-2 phosphorylation to apoptosis induction in human B-cells is dependent on the mode of action of specific stresses
- PMID: 18201741
- PMCID: PMC2349100
- DOI: 10.1016/j.taap.2007.11.032
The contribution of c-Jun N-terminal kinase activation and subsequent Bcl-2 phosphorylation to apoptosis induction in human B-cells is dependent on the mode of action of specific stresses
Abstract
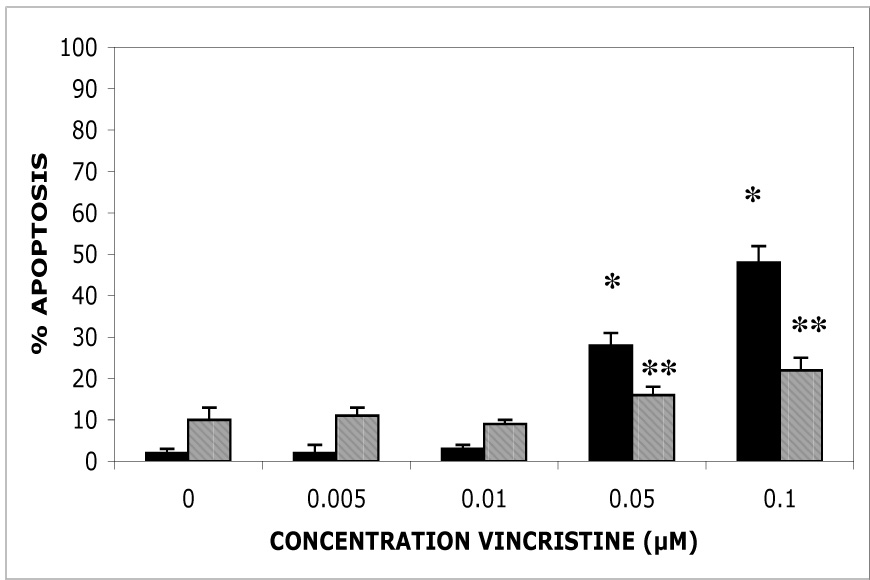
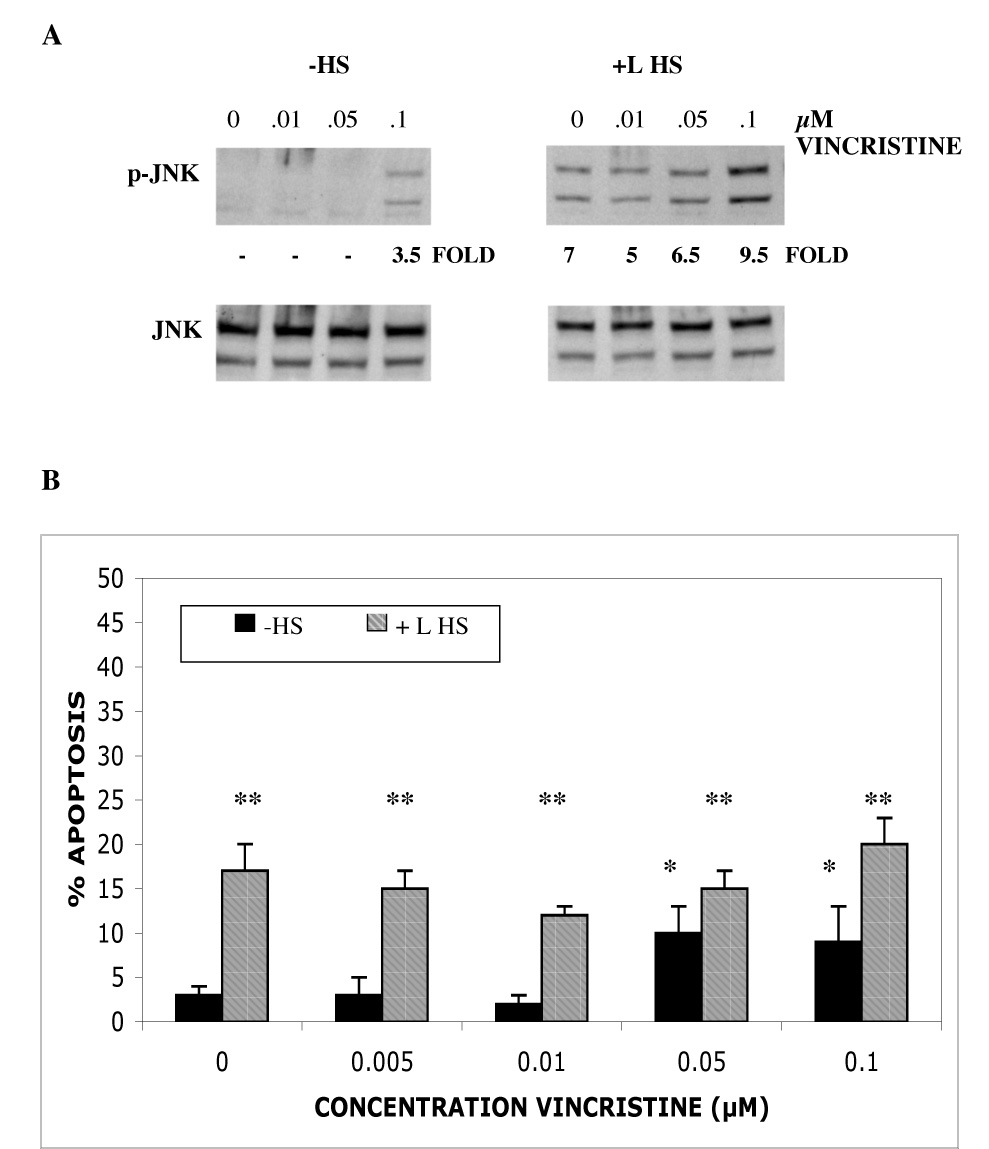
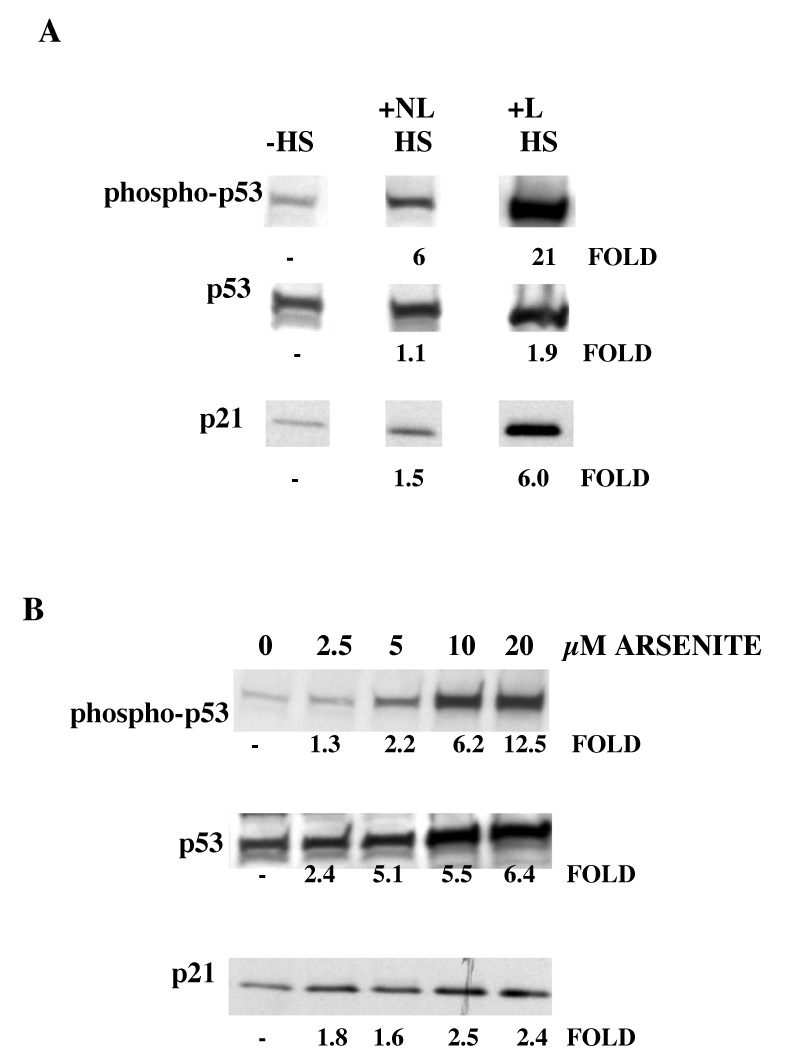
The c-Jun N-terminal kinase (JNK) pathway can play paradoxical roles as either a pro-survival or a pro-cell death pathway depending on type of stress and cell type. The goal of the present study was to determine the role of JNK pathway signaling for regulating B-cell apoptosis in two important but contrasting situations--global proteotoxic damage, induced by arsenite and hyperthermia, versus specific microtubule inhibition, induced by the anti-cancer drug vincristine, using the EW36 B-cell line. This cell line over-expresses the Bcl-2 protein and is a useful model to identify treatments that can overcome multi-drug resistance in lymphoid cells. Exposure of EW36 B-cells to arsenite or lethal hyperthermia resulted in activation of the JNK pathway and induction of apoptosis. However, pharmacological inhibition of the JNK pathway did not inhibit apoptosis, indicating that JNK pathway activation is not required for apoptosis induction by these treatments. In contrast, vincristine treatment of EW36 B-cells resulted in JNK activation and apoptosis that was suppressed by JNK inhibition. A critical difference between the two types of stress treatments was that only vincristine-induced JNK activation resulted in phosphorylation of Bcl-2 at threonine-56, a modification that can block its anti-apoptotic function. Importantly, Bcl-2 phosphorylation was attenuated by JNK inhibition implicating JNK as the upstream kinase. Furthermore, arsenite and hyperthermia treatments activated a p53/p21 pathway associated with apoptosis induction, whereas vincristine did not activate this pathway. These results reveal two stress-activated pathways, one JNK-dependent and another JNK-independent, either of which can bypass Bcl-2 mediated resistance, resulting in cell death.
Figures







References
-
- Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. - PubMed
-
- Bloom SE, Lemley AT, Muscarella DE. Potentiation of apoptosis by heat stress plus pesticide exposure in stress resistant human B-lymphoma cells and its attenuation through interaction with follicular dendritic cells: role for c-Jun N-terminal kinase signaling. Toxicol. Sci. 2006;89:214–223. - PubMed
-
- Chen Y-R, Wang X, Templeton D, Davis RJ, Tan T-H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. J. Biol.Chem. 1996;271:31929–31936. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
