

Sensing of viral infection and activation of innate immunity by toll-like receptor 3
- PMID: 18202435
- PMCID: PMC2223843
- DOI: 10.1128/CMR.00022-07
Sensing of viral infection and activation of innate immunity by toll-like receptor 3
Abstract
Toll-like receptors (TLRs) form a major group of transmembrane receptors that are involved in the detection of invading pathogens. Double-stranded RNA is a marker for viral infection that is recognized by TLR3. TLR3 triggering activates specific signaling pathways that culminate in the activation of NF-kappaB and IRF3 transcription factors, as well as apoptosis, enabling the host to mount an effective innate immune response through the induction of cytokines, chemokines, and other proinflammatory mediators. In this review, we describe the paradoxical role of TLR3 in innate immunity against different viruses and in viral pathogenesis but also the evidence for TLR3 as a "danger" receptor in nonviral diseases. We also discuss the structure and cellular localization of TLR3, as well as the complex signaling and regulatory events that contribute to TLR3-mediated immune responses.
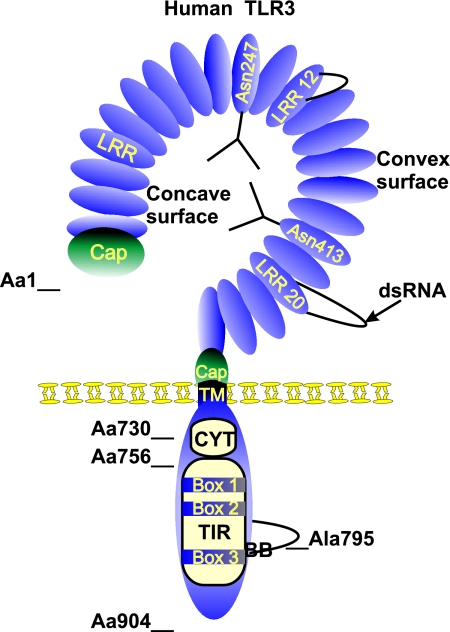
Figures


References
-
- Aksoy, E., W. Vanden Berghe, S. Detienne, Z. Amraoui, K. A. Fitzgerald, G. Haegeman, M. Goldman, and F. Willems. 2005. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-κB activation and IFN-β synthesis downstream of Toll-like receptor 3 and 4. Eur. J. Immunol. 35:2200-2209. - PubMed
-
- Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413:732-738. - PubMed
-
- An, H., W. Zhao, J. Hou, Y. Zhang, Y. Xie, Y. Zheng, H. Xu, C. Qian, J. Zhou, Y. Yu, S. Liu, G. Feng, and X. Cao. 2006. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity 25:919-928. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
