

Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion
- PMID: 18202440
- PMCID: PMC2223840
- DOI: 10.1128/CMR.00032-07
Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion
Abstract
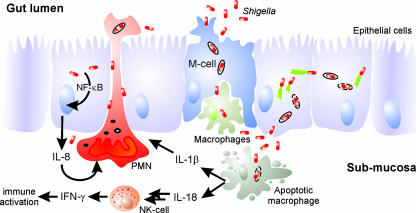
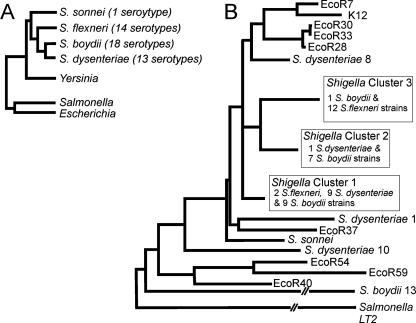
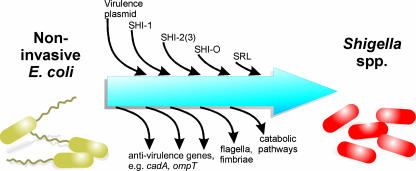
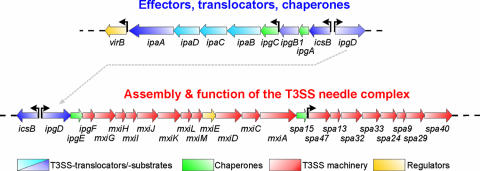
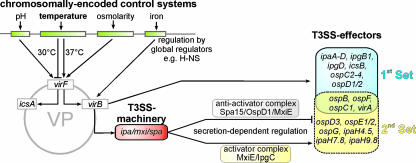
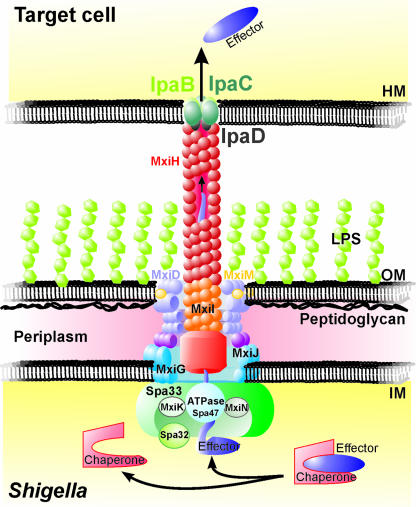
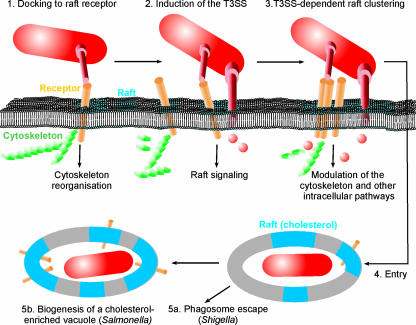
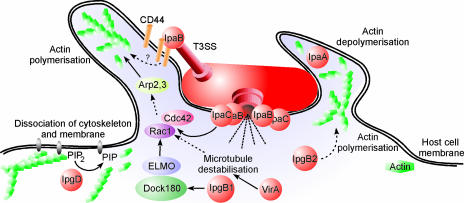
Shigella spp. are gram-negative pathogenic bacteria that evolved from harmless enterobacterial relatives and may cause devastating diarrhea upon ingestion. Research performed over the last 25 years revealed that a type III secretion system (T3SS) encoded on a large plasmid is a key virulence factor of Shigella flexneri. The T3SS determines the interactions of S. flexneri with intestinal cells by consecutively translocating two sets of effector proteins into the target cells. Thus, S. flexneri controls invasion into EC, intra- and intercellular spread, macrophage cell death, as well as host inflammatory responses. Some of the translocated effector proteins show novel biochemical activities by which they intercept host cell signal transduction pathways. An understanding of the molecular mechanisms underlying Shigella pathogenesis will foster the development of a safe and efficient vaccine, which, in parallel with improved hygiene, should curb infections by this widespread pathogen.
Figures
References
-
- Abe, A., T. Matsuzawa, and A. Kuwae. 2005. Type-III effectors: sophisticated bacterial virulence factors. C. R. Biol. 328:413-428. - PubMed
-
- Adler, B., C. Sasakawa, T. Tobe, S. Makino, K. Komatsu, and M. Yoshikawa. 1989. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 3:627-635. - PubMed
-
- Akeda, Y., and J. E. Galan. 2005. Chaperone release and unfolding of substrates in type III secretion. Nature 437:911-915. - PubMed
-
- Alder, N. N., and S. M. Theg. 2003. Energy use by biological protein transport pathways. Trends Biochem. Sci. 28:442-451. - PubMed
-
- Al-Hasani, K., I. R. Henderson, H. Sakellaris, K. Rajakumar, T. Grant, J. P. Nataro, R. Robins-Browne, and B. Adler. 2000. The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect. Immun. 68:2457-2463. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
