

Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis
- PMID: 18204489
- PMCID: PMC2268064
- DOI: 10.1038/sj.bjp.0707659
Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis
Abstract
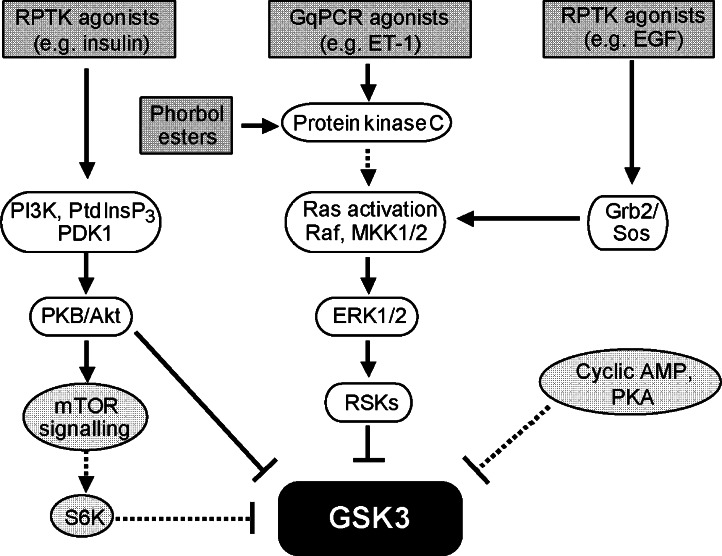
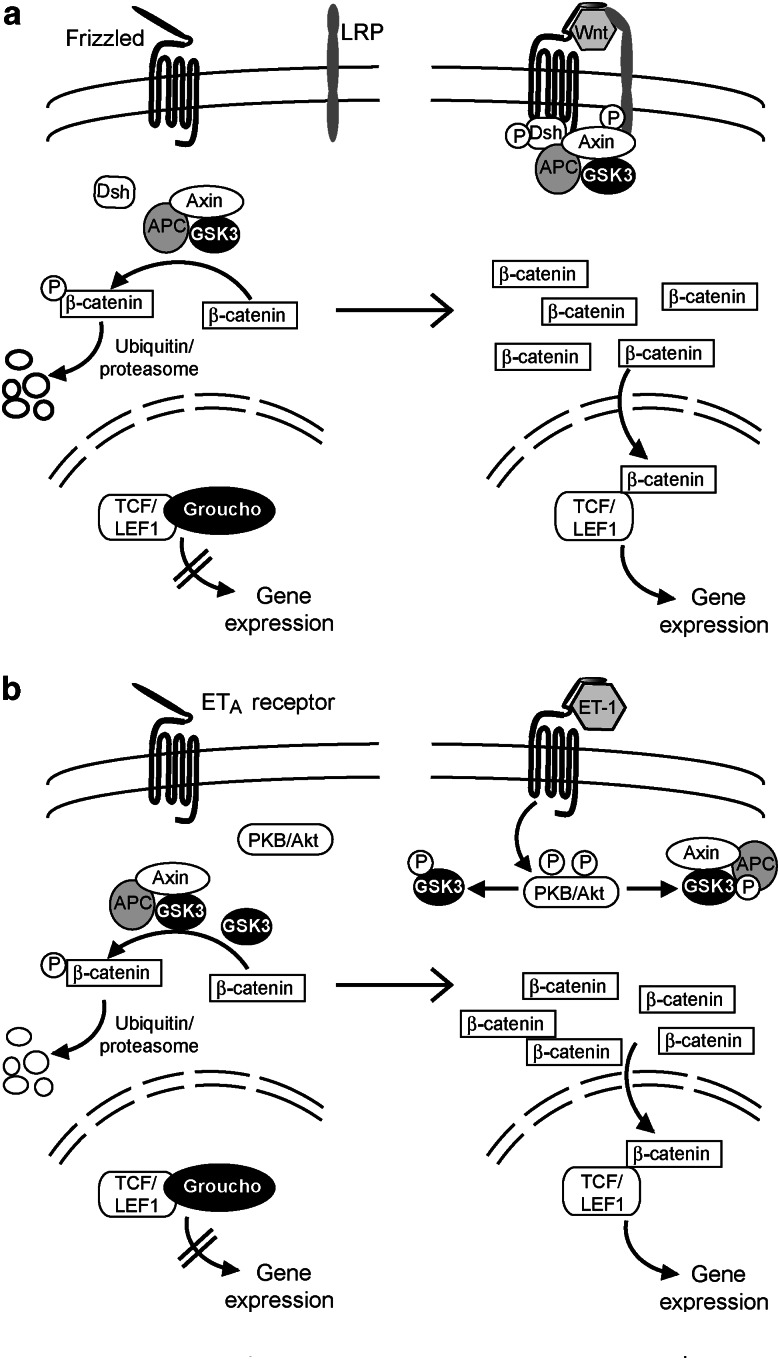
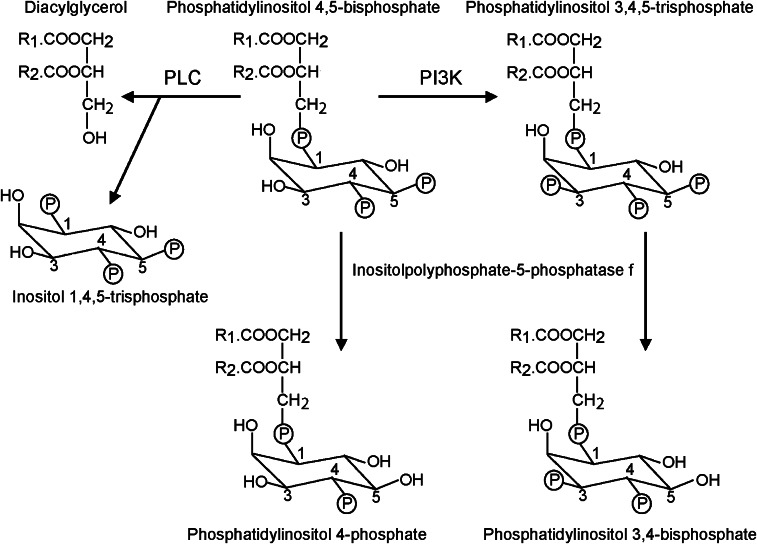
Glycogen synthase kinase 3 (GSK3, of which there are two isoforms, GSK3alpha and GSK3beta) was originally characterized in the context of regulation of glycogen metabolism, though it is now known to regulate many other cellular processes. Phosphorylation of GSK3alpha(Ser21) and GSK3beta(Ser9) inhibits their activity. In the heart, emphasis has been placed particularly on GSK3beta, rather than GSK3alpha. Importantly, catalytically-active GSK3 generally restrains gene expression and, in the heart, catalytically-active GSK3 has been implicated in anti-hypertrophic signalling. Inhibition of GSK3 results in changes in the activities of transcription and translation factors in the heart and promotes hypertrophic responses, and it is generally assumed that signal transduction from hypertrophic stimuli to GSK3 passes primarily through protein kinase B/Akt (PKB/Akt). However, recent data suggest that the situation is far more complex. We review evidence pertaining to the role of GSK3 in the myocardium and discuss effects of genetic manipulation of GSK3 activity in vivo. We also discuss the signalling pathways potentially regulating GSK3 activity and propose that, depending on the stimulus, phosphorylation of GSK3 is independent of PKB/Akt. Potential GSK3 substrates studied in relation to myocardial hypertrophy include nuclear factors of activated T cells, beta-catenin, GATA4, myocardin, CREB, and eukaryotic initiation factor 2Bvarepsilon. These and other transcription factor substrates putatively important in the heart are considered. We discuss whether cardiac pathologies could be treated by therapeutic intervention at the GSK3 level but conclude that any intervention would be premature without greater understanding of the precise role of GSK3 in cardiac processes.
Figures


References
-
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor–Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. - PubMed
-
- Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. - PubMed
-
- Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC, Leinwand LA. Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J Appl Physiol. 2001;90:1900–1908. - PubMed
-
- Andersen JB, Rourke BC, Caiozzo VJ, Bennett AF, Hicks JW. Postprandial hypertrophy in pythons. Nature. 2005;434:37–38. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
