

Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation
- PMID: 18206965
- PMCID: PMC2234035
- DOI: 10.1016/j.molcel.2007.11.031
Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation
Abstract
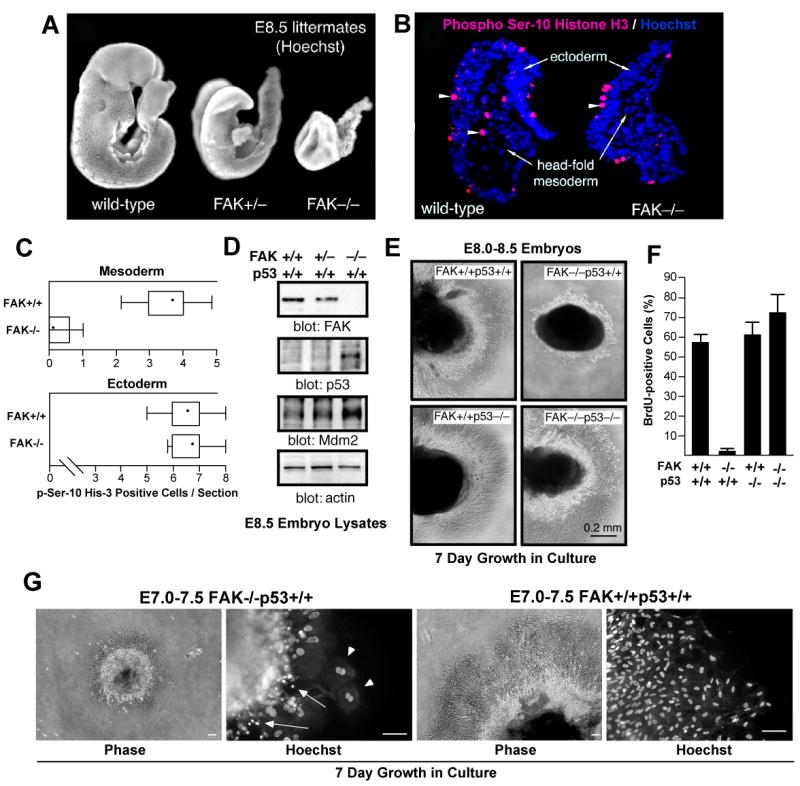
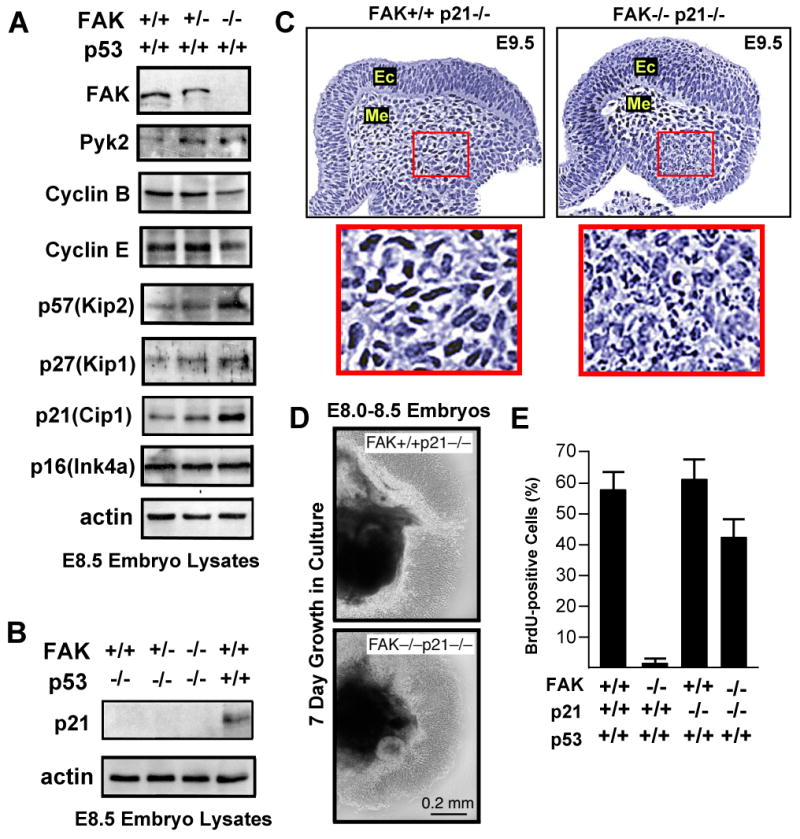
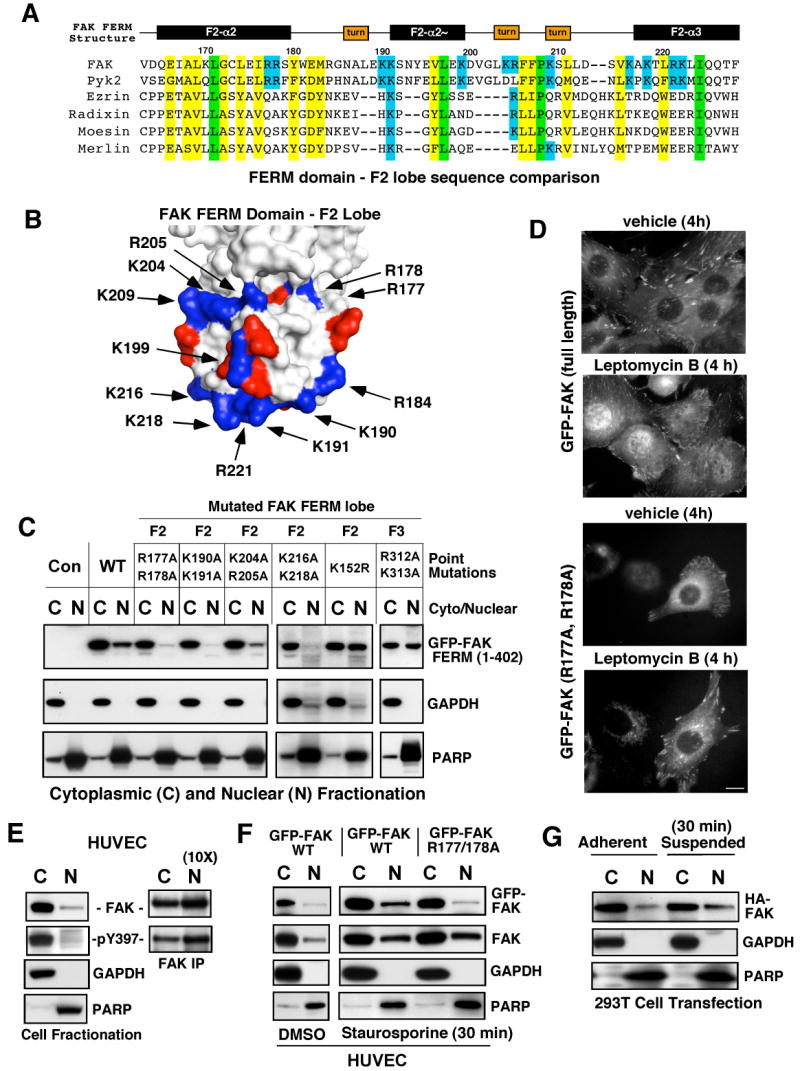
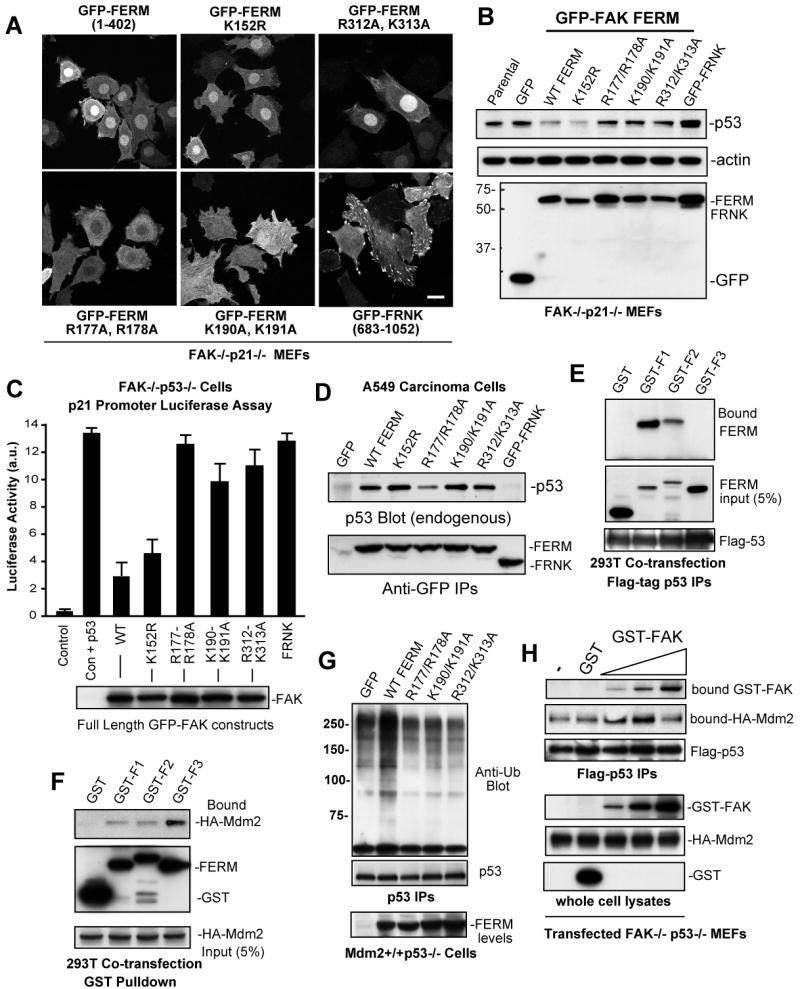
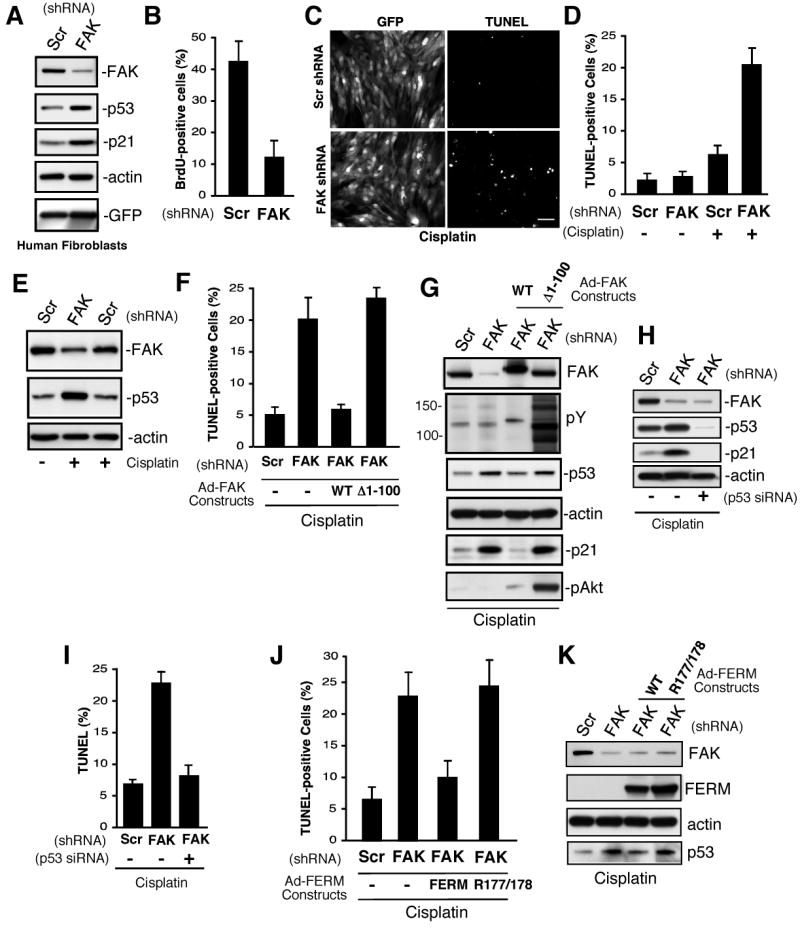
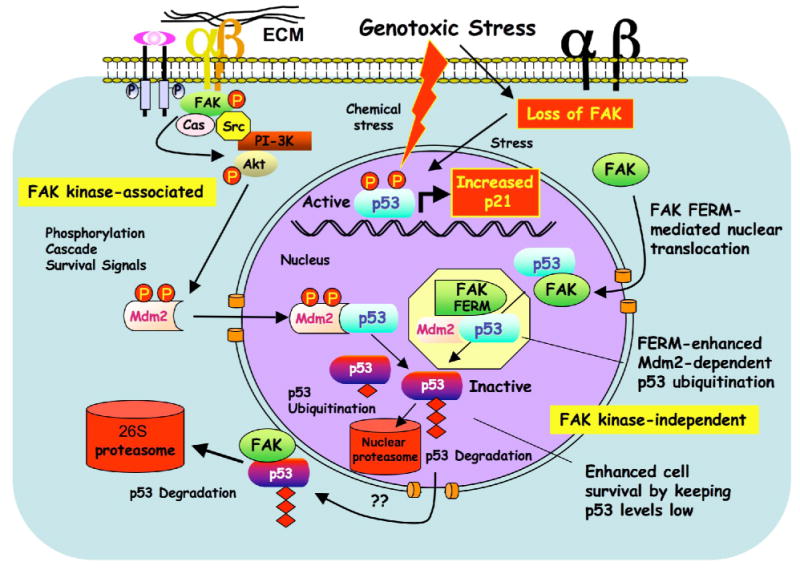
FAK is known as an integrin- and growth factor-associated tyrosine kinase promoting cell motility. Here we show that, during mouse development, FAK inactivation results in p53- and p21-dependent mesodermal cell growth arrest. Reconstitution of primary FAK-/-p21-/- fibroblasts revealed that FAK, in a kinase-independent manner, facilitates p53 turnover via enhanced Mdm2-dependent p53 ubiquitination. p53 inactivation by FAK required FAK FERM F1 lobe binding to p53, FERM F2 lobe-mediated nuclear localization, and FERM F3 lobe for connections to Mdm2 and proteasomal degradation. Staurosporine or loss of cell adhesion enhanced FERM-dependent FAK nuclear accumulation. In primary human cells, FAK knockdown raised p53-p21 levels and slowed cell proliferation but did not cause apoptosis. Notably, FAK knockdown plus cisplatin triggered p53-dependent cell apoptosis, which was rescued by either full-length FAK or FAK FERM re-expression. These studies define a scaffolding role for nuclear FAK in facilitating cell survival through enhanced p53 degradation under conditions of cellular stress.
Figures

References
-
- Cohen LA, Guan JL. Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J Biol Chem. 2005;280:8197–8207. - PubMed
-
- Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
