

Progress in neuroprotective strategies for preventing epilepsy
- PMID: 18207302
- PMCID: PMC2441599
- DOI: 10.1016/j.pneurobio.2007.10.010
Progress in neuroprotective strategies for preventing epilepsy
Abstract
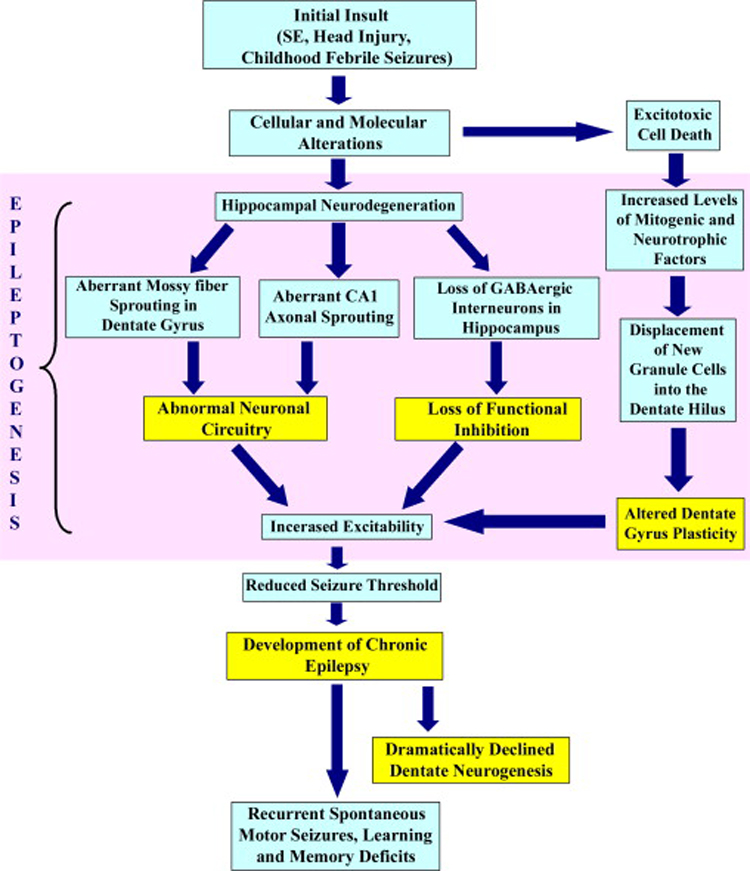
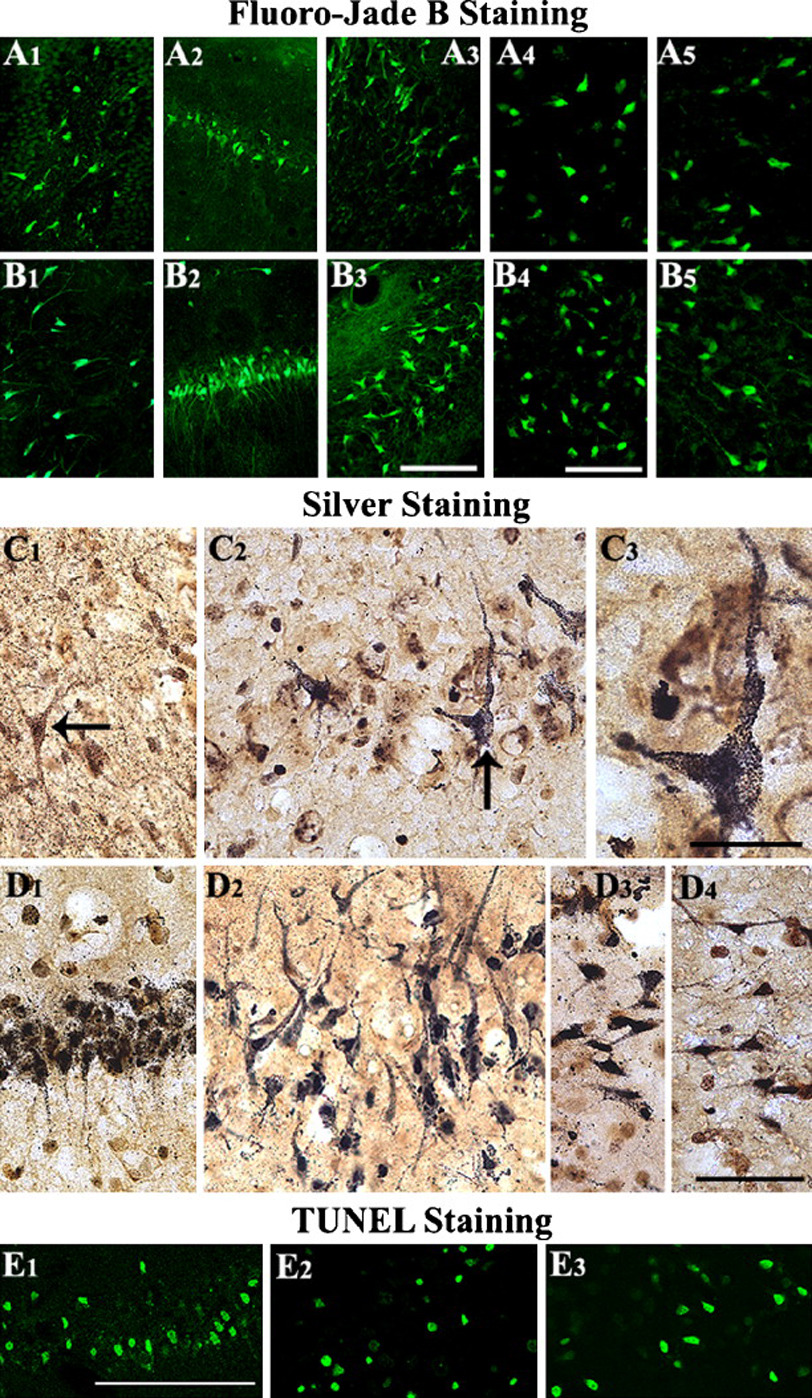
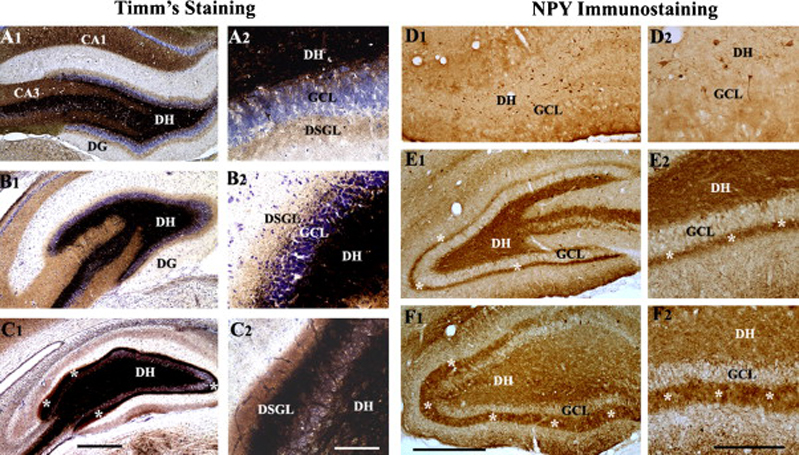
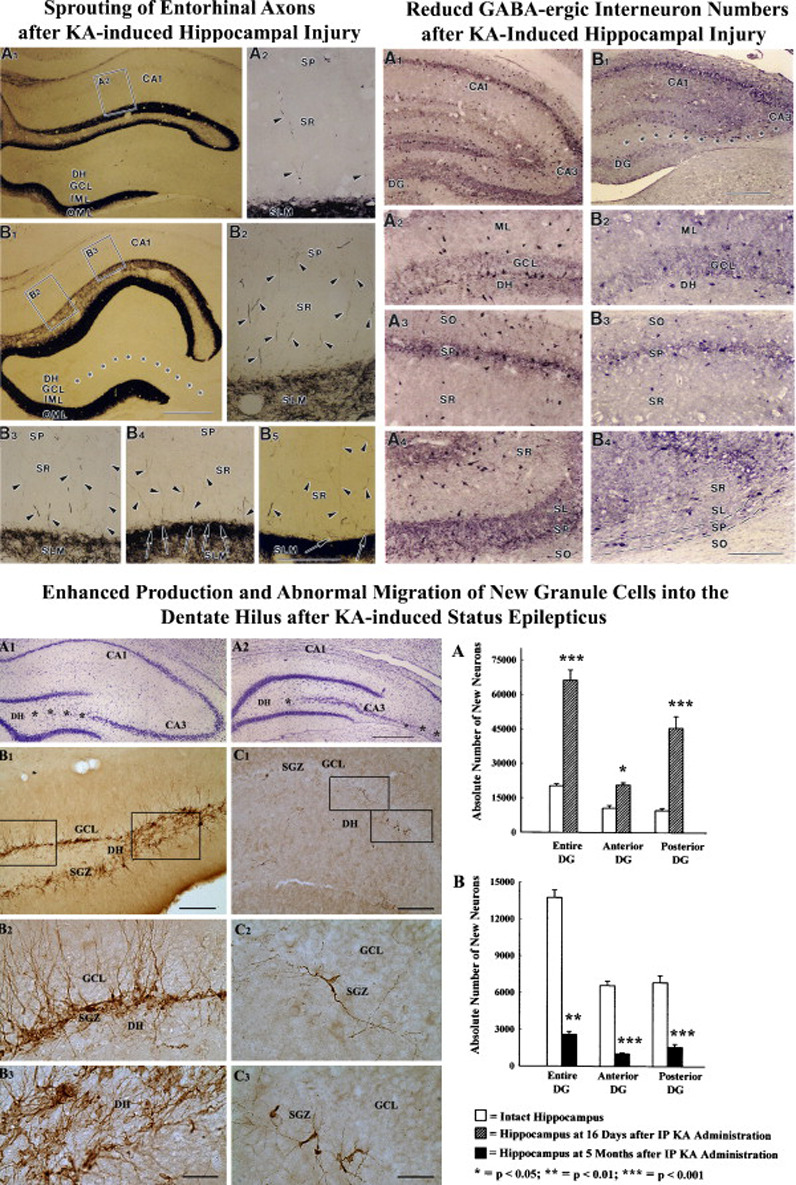
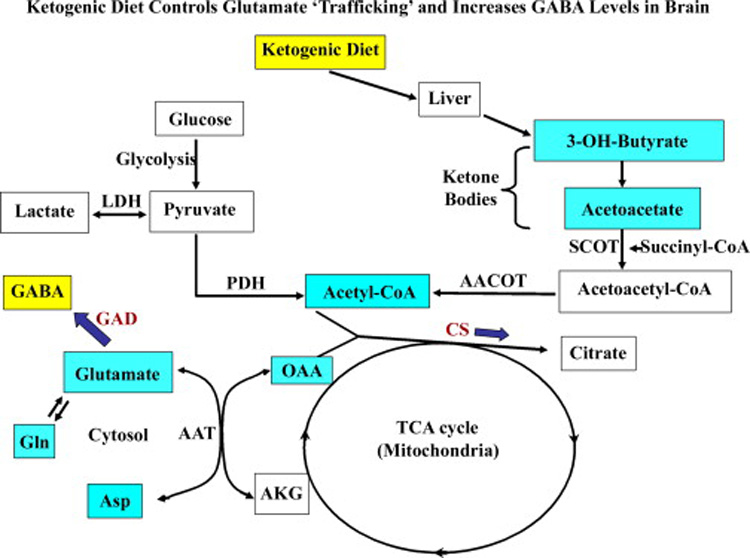
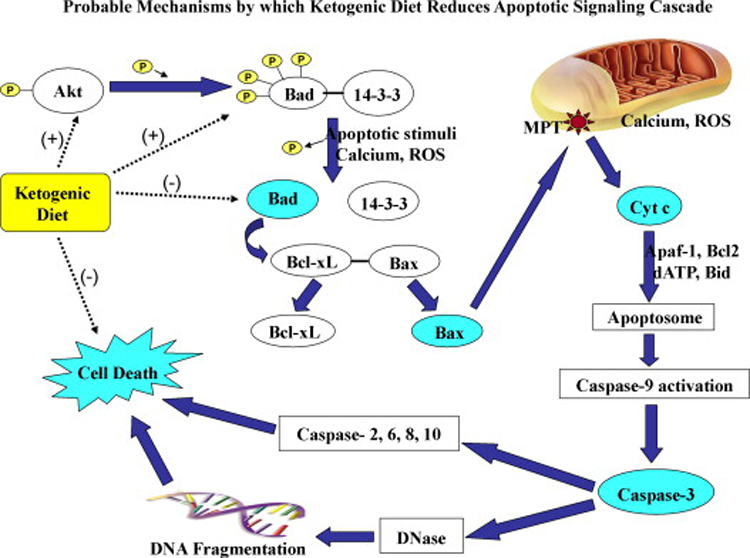
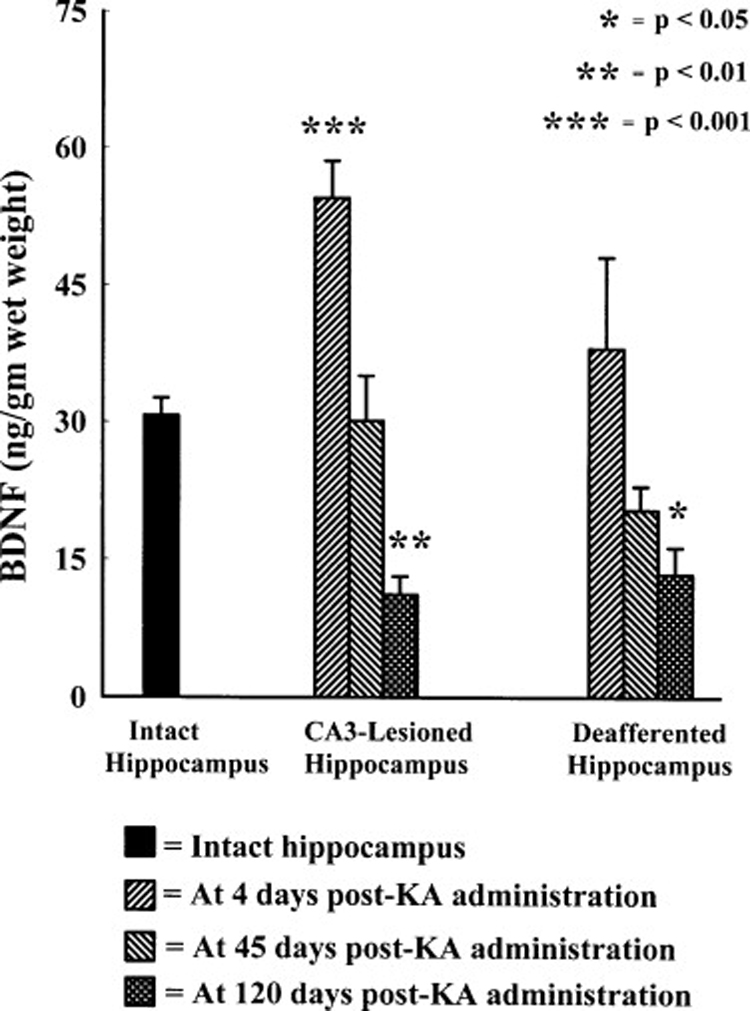
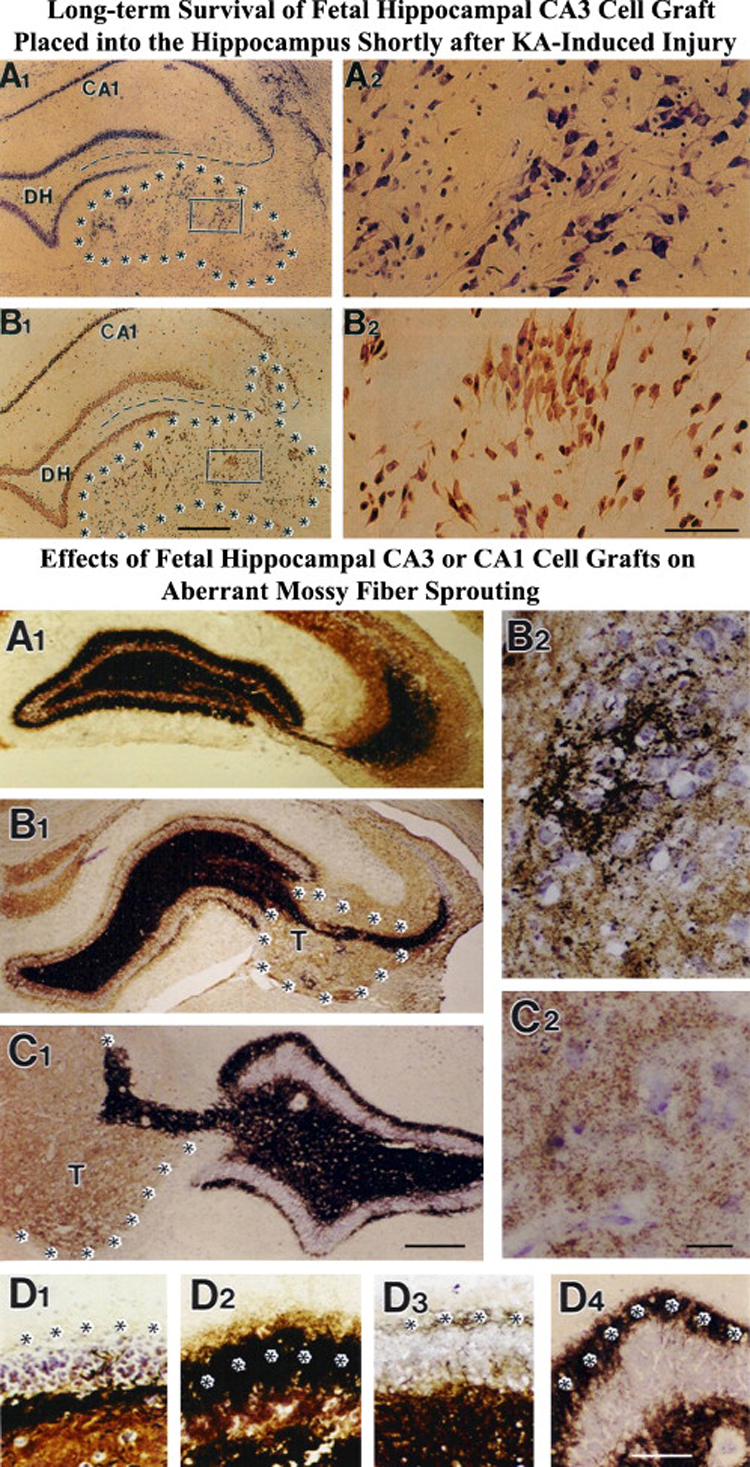
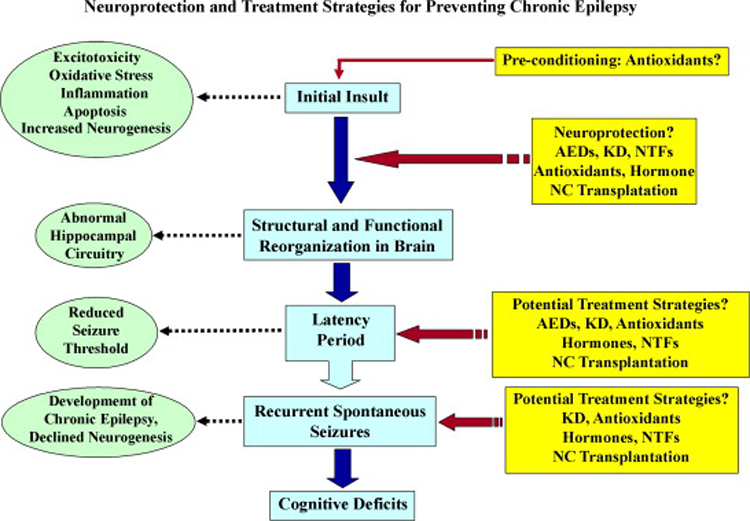
Neuroprotection is increasingly considered as a promising therapy for preventing and treating temporal lobe epilepsy (TLE). The development of chronic TLE, also termed as epileptogenesis, is a dynamic process. An initial precipitating injury (IPI) such as the status epilepticus (SE) leads to neurodegeneration, abnormal reorganization of the brain circuitry and a significant loss of functional inhibition. All of these changes likely contribute to the development of chronic epilepsy, characterized by spontaneous recurrent motor seizures (SRMS) and learning and memory deficits. The purpose of this review is to discuss the current state of knowledge pertaining to neuroprotection in epileptic conditions, and to highlight the efficacy of distinct neuroprotective strategies for preventing or treating chronic TLE. Although the administration of certain conventional and new generation anti-epileptic drugs is effective for primary neuroprotection such as reduced neurodegeneration after acute seizures or the SE, their competence for preventing the development of chronic epilepsy after an IPI is either unknown or not promising. On the other hand, alternative strategies such as the ketogenic diet therapy, administration of distinct neurotrophic factors, hormones or antioxidants seem useful for preventing and treating chronic TLE. However, long-term studies on the efficacy of these approaches introduced at different time-points after the SE or an IPI are lacking. Additionally, grafting of fetal hippocampal cells at early time-points after an IPI holds considerable promise for preventing TLE, though issues regarding availability of donor cells, ethical concerns, timing of grafting after SE, and durability of graft-mediated seizure suppression need to be resolved for further advances with this approach. Overall, from the studies performed so far, there is consensus that neuroprotective strategies need to be employed as quickly as possible after the onset of the SE or an IPI for considerable beneficial effects. Nevertheless, ideal strategies that are capable of facilitating repair and functional recovery of the brain after an IPI and preventing the evolution of IPI into chronic epilepsy are still hard to pin down.
Figures
References
-
- Acharya MM, Katyare SS. Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp Neurol. 2005;192:79–88. - PubMed
-
- Acuna-Castroviejo D, Escames G, Macias M, Munoz Hoyos A, Molina Carballo A, Arauzo M, Montes R. Cell protective role of melatonin in the brain. J Pineal Res. 1995;19:57–63. - PubMed
-
- Adams B, Sazgar M, Osehobo P, Van der Zee CE, Diamond J, Fahnestock M, Racine RJ. Nerve growth factor accelerates seizure development, enhances mossy fiber sprouting, and attenuates seizure-induced decreases in neuronal density in the kindling model of epilepsy. J Neurosci. 1997;17:5288–5296. - PMC - PubMed
-
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature Rev. 2002;3:383–394. - PubMed
-
- Ajmone-Cat MA, Iosif RE, Ekdahl CT, Kokaia Z, Minghetti L, Lindvall O. Prostaglandin E2 and BDNF levels in rat hippocampus are negatively correlated with status epilepticus severity: no impact on survival of seizure-generated neurons. Neurobiol Dis. 2006;23:23–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
