

Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression
- PMID: 18212064
- PMCID: PMC2268392
- DOI: 10.1128/MCB.01442-07
Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression
Abstract
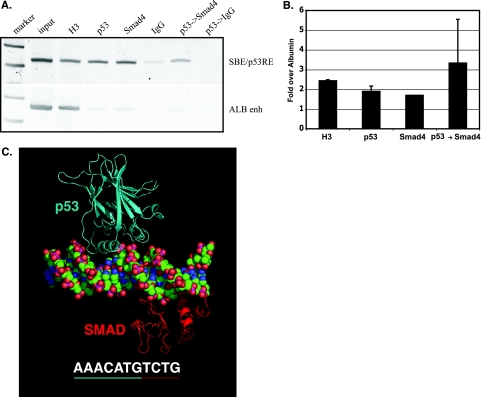
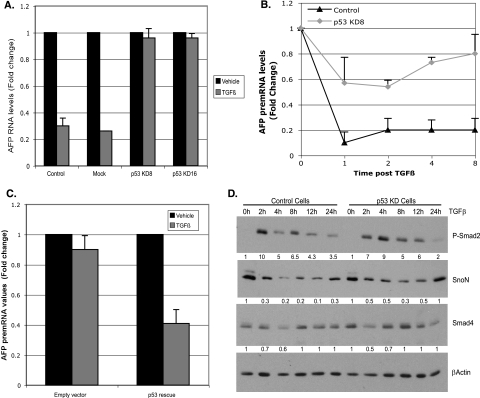
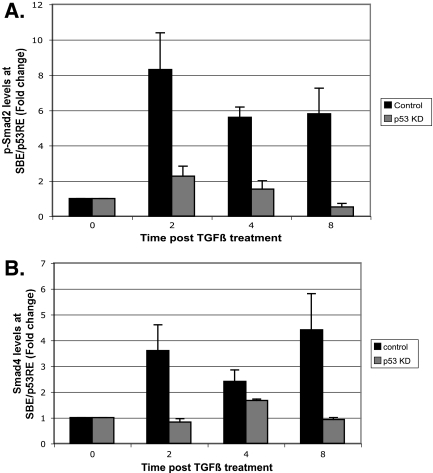
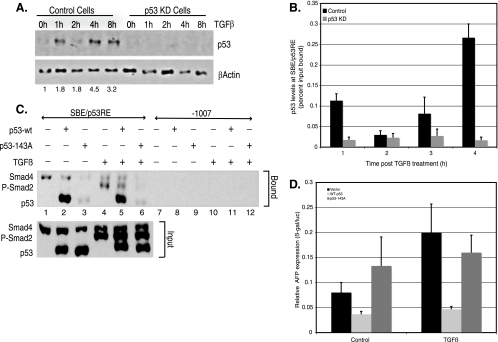
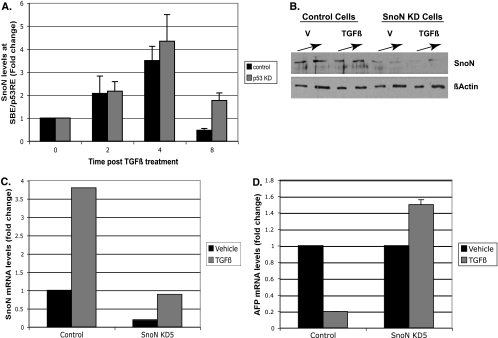
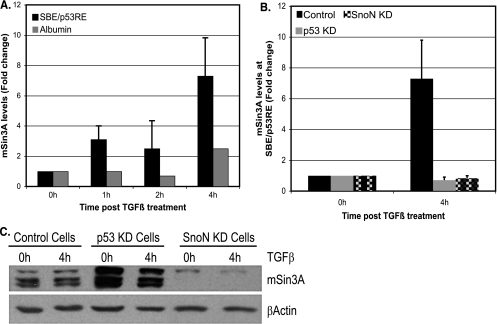
In hepatic cells, Smad and SnoN proteins converge with p53 to repress transcription of AFP, an oncodevelopmental tumor marker aberrantly reactivated in hepatoma cells. Using p53- and SnoN-depleted hepatoma cell clones, we define a mechanism for repression mediated by this novel transcriptional partnership. We find that p53 anchors activated Smads and the corepressor mSin3A to the AFP distal promoter. Sequential chromatin immunoprecipitation analyses and molecular modeling indicate that p53 and Smad proteins simultaneously occupy overlapping p53 and Smad regulatory elements to establish repression of AFP transcription. In addition to its well-known function in antagonizing transforming growth factor beta (TGF-beta) responses, we find that SnoN actively participates in AFP repression by positively regulating mSin3A protein levels. We propose that activation of TGF-beta signaling restores a dynamic interplay between p53 and TGF-beta effectors that cooperate to effectively target mSin3A to tumor marker AFP and reestablish transcription repression.
Figures
Similar articles
-
A direct intersection between p53 and transforming growth factor beta pathways targets chromatin modification and transcription repression of the alpha-fetoprotein gene.Mol Cell Biol. 2005 Feb;25(3):1200-12. doi: 10.1128/MCB.25.3.1200-1212.2005. Mol Cell Biol. 2005. PMID: 15657445 Free PMC article.
-
Repression of p53-mediated transcription by adenovirus E1B 55-kDa does not require corepressor mSin3A and histone deacetylases.J Biol Chem. 2007 Mar 9;282(10):7001-10. doi: 10.1074/jbc.M610749200. Epub 2007 Jan 5. J Biol Chem. 2007. PMID: 17209038
-
KLF11 mediates a critical mechanism in TGF-beta signaling that is inactivated by Erk-MAPK in pancreatic cancer cells.Gastroenterology. 2004 Aug;127(2):607-20. doi: 10.1053/j.gastro.2004.05.018. Gastroenterology. 2004. PMID: 15300592
-
Repression of TGF-beta signaling by the oncogenic protein SKI in human melanomas: consequences for proliferation, survival, and metastasis.Oncogene. 2003 May 19;22(20):3123-9. doi: 10.1038/sj.onc.1206452. Oncogene. 2003. PMID: 12793438 Review.
-
Transcriptional Control by the SMADs.Cold Spring Harb Perspect Biol. 2016 Oct 3;8(10):a022079. doi: 10.1101/cshperspect.a022079. Cold Spring Harb Perspect Biol. 2016. PMID: 27449814 Free PMC article. Review.
Cited by
-
Microvascular significance of TGF-β axis activation in COVID-19.Front Cardiovasc Med. 2023 Jan 6;9:1054690. doi: 10.3389/fcvm.2022.1054690. eCollection 2022. Front Cardiovasc Med. 2023. PMID: 36684608 Free PMC article. Review.
-
The current state of chromatin immunoprecipitation.Mol Biotechnol. 2010 May;45(1):87-100. doi: 10.1007/s12033-009-9239-8. Mol Biotechnol. 2010. PMID: 20077036 Review.
-
Contextual Regulation of TGF-β Signaling in Liver Cancer.Cells. 2019 Oct 11;8(10):1235. doi: 10.3390/cells8101235. Cells. 2019. PMID: 31614569 Free PMC article. Review.
-
One function--multiple mechanisms: the manifold activities of p53 as a transcriptional repressor.J Biomed Biotechnol. 2011;2011:464916. doi: 10.1155/2011/464916. Epub 2011 Mar 8. J Biomed Biotechnol. 2011. PMID: 21436991 Free PMC article. Review.
-
Rescuing lung development through embryonic inhibition of histone acetylation.Sci Transl Med. 2024 Jan 31;16(732):eadc8930. doi: 10.1126/scitranslmed.adc8930. Epub 2024 Jan 31. Sci Transl Med. 2024. PMID: 38295182 Free PMC article.
References
-
- Appella, E., and C. W. Anderson. 2000. Signaling to p53: breaking the posttranslational modification code. Pathol. Biol. (Paris) 48227-245. - PubMed
-
- Arva, N. C., T. R. Gopen, K. E. Talbott, L. E. Campbell, A. Chicas, D. E. White, G. L. Bond, A. J. Levine, and J. Bargonetti. 2005. A chromatin-associated and transcriptionally inactive p53-Mdm2 complex occurs in mdm2 SNP309 homozygous cells. J. Biol. Chem. 28026776-26787. - PubMed
-
- Bakker, J., C. De Mees, Q. Douhard, J. Balthazart, P. Gabant, J. Szpirer, and C. Szpirer. 2006. Alpha-fetoprotein protects the developing female mouse brain from masculinization and defeminization by estrogens. Nat. Neurosci. 9220-226. - PubMed
-
- Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D 54905-921. - PubMed
-
- Cordenonsi, M., S. Dupont, S. Maretto, A. Insinga, C. Imbriano, and S. Piccolo. 2003. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 113301-314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous