

Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma
- PMID: 18215105
- PMCID: PMC2211560
- DOI: 10.1371/journal.pmed.0050008
Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma
Abstract
Background: There is much discussion in the cancer drug development community about how to incorporate molecular tools into early-stage clinical trials to assess target modulation, measure anti-tumor activity, and enrich the clinical trial population for patients who are more likely to benefit. Small, molecularly focused clinical studies offer the promise of the early definition of optimal biologic dose and patient population.
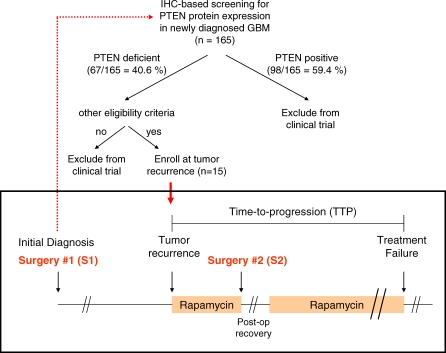
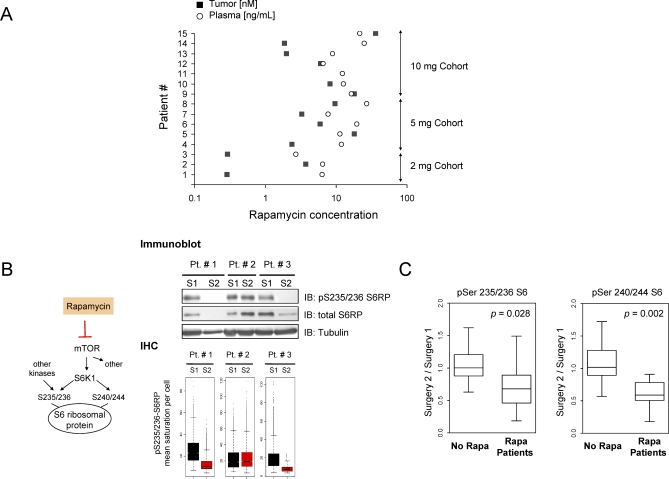
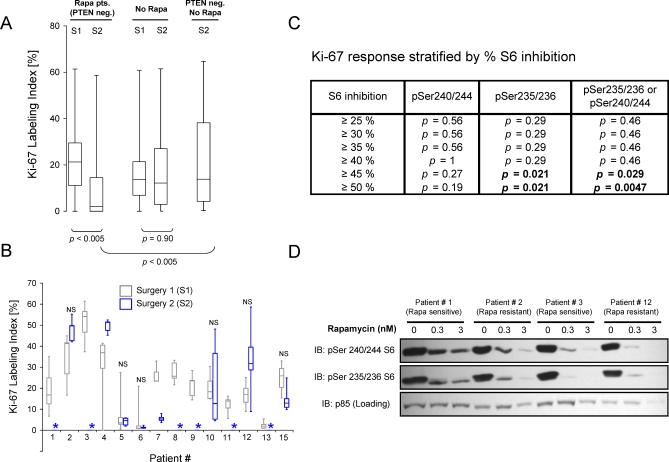
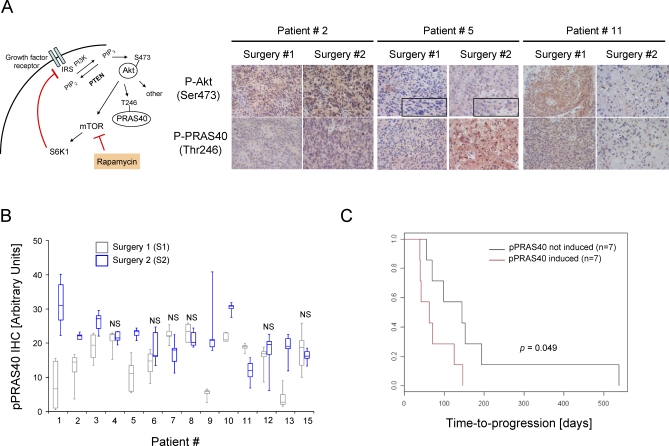
Methods and findings: Based on preclinical evidence that phosphatase and tensin homolog deleted on Chromosome 10 (PTEN) loss sensitizes tumors to the inhibition of mammalian target of rapamycin (mTOR), we conducted a proof-of-concept Phase I neoadjuvant trial of rapamycin in patients with recurrent glioblastoma, whose tumors lacked expression of the tumor suppressor PTEN. We aimed to assess the safety profile of daily rapamycin in patients with glioma, define the dose of rapamycin required for mTOR inhibition in tumor tissue, and evaluate the antiproliferative activity of rapamycin in PTEN-deficient glioblastoma. Although intratumoral rapamycin concentrations that were sufficient to inhibit mTOR in vitro were achieved in all patients, the magnitude of mTOR inhibition in tumor cells (measured by reduced ribosomal S6 protein phosphorylation) varied substantially. Tumor cell proliferation (measured by Ki-67 staining) was dramatically reduced in seven of 14 patients after 1 wk of rapamycin treatment and was associated with the magnitude of mTOR inhibition (p = 0.0047, Fisher exact test) but not the intratumoral rapamycin concentration. Tumor cells harvested from the Ki-67 nonresponders retained sensitivity to rapamycin ex vivo, indicating that clinical resistance to biochemical mTOR inhibition was not cell-intrinsic. Rapamycin treatment led to Akt activation in seven patients, presumably due to loss of negative feedback, and this activation was associated with shorter time-to-progression during post-surgical maintenance rapamycin therapy (p < 0.05, Logrank test).
Conclusions: Rapamycin has anticancer activity in PTEN-deficient glioblastoma and warrants further clinical study alone or in combination with PI3K pathway inhibitors. The short-term treatment endpoints used in this neoadjuvant trial design identified the importance of monitoring target inhibition and negative feedback to guide future clinical development.
Trial registration: http://www.ClinicalTrials.gov (#NCT00047073).
Conflict of interest statement
Figures
Comment in
-
Incorporating molecular tools into early-stage clinical trials.PLoS Med. 2008 Jan 22;5(1):e21. doi: 10.1371/journal.pmed.0050021. PLoS Med. 2008. PMID: 18215108 Free PMC article. Review.
References
-
- Arteaga CL, Baselga J. Tyrosine kinase inhibitors: why does the current process of clinical development not apply to them? Cancer Cell. 2004;5:525–531. - PubMed
-
- Sarker D, Workman P. Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res. 2007;96:213–268. - PubMed
-
- Michaelis LC, Ratain MJ. Measuring response in a post-RECIST world: from black and white to shades of grey. Nat Rev Cancer. 2006;6:409–414. - PubMed
-
- Smith IE, Walsh G, Skene A, Llombart A, Mayordomo JI, et al. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol. 2007;25:3816–3822. - PubMed
-
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. 2007;99:167–170. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
