

Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance
- PMID: 18216219
- PMCID: PMC2528847
- DOI: 10.1210/er.2006-0045
Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance
Abstract
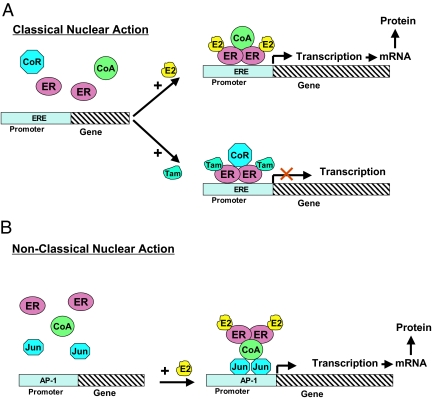
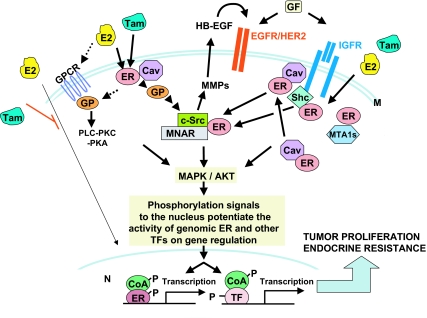
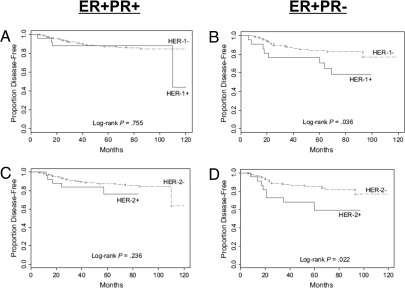
Breast cancer evolution and tumor progression are governed by the complex interactions between steroid receptor [estrogen receptor (ER) and progesterone receptor] and growth factor receptor signaling. In recent years, the field of cancer therapy has witnessed the emergence of multiple strategies targeting these specific cancer pathways and key molecules (ER and growth factor receptors) to arrest tumor growth and achieve tumor eradication; treatment success, however, has varied and both de novo (up front) and acquired resistance have proven a challenge. Recent studies of ER biology have revealed new insights into ER action in breast cancer and have highlighted the role of an intimate crosstalk between the ER and HER family signaling pathways as a fundamental contributor to the development of resistance to endocrine therapies against the ER pathway. The aim of this review article is to summarize the current knowledge on mechanisms of resistance of breast cancer cells to endocrine therapies due to the crosstalk between the ER and the HER growth factor receptor signaling pathways and to explore new available therapeutic strategies that could prolong duration of response and circumvent endocrine resistant tumor growth.
Figures
References
-
- Group EBCTC 1998 Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351:1451–1467 - PubMed
-
- Gradishar WJ 2004 Tamoxifen—what next? Oncologist 9:378–384 - PubMed
-
- Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N 1998 Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 90:1371–1388 - PubMed
-
- Osborne CK, Schiff R 2005 Aromatase inhibitors: future directions. J Steroid Biochem Mol Biol 95:183–187 - PubMed
-
- Johnston SR, Head J, Pancholi S, Detre S, Martin LA, Smith IE, Dowsett M 2003 Integration of signal transduction inhibitors with endocrine therapy: an approach to overcoming hormone resistance in breast cancer. Clin Cancer Res 9:524S–532S - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
