

Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease
- PMID: 18218341
- PMCID: PMC3107703
- DOI: 10.1016/j.molmed.2007.12.002
Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease
Abstract
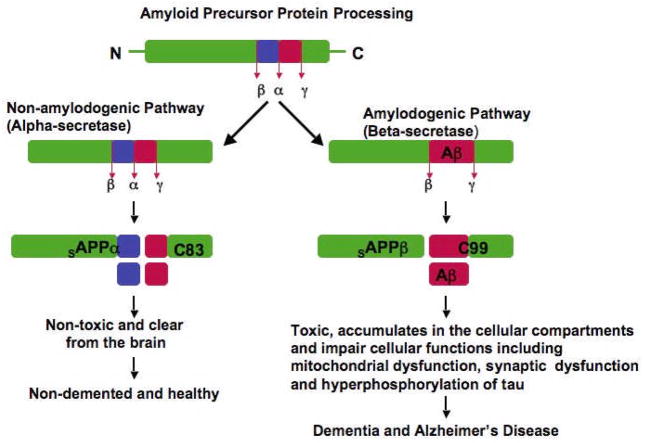
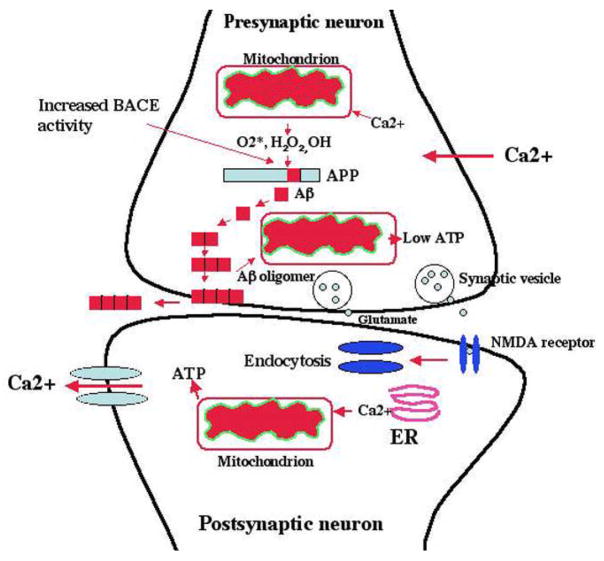
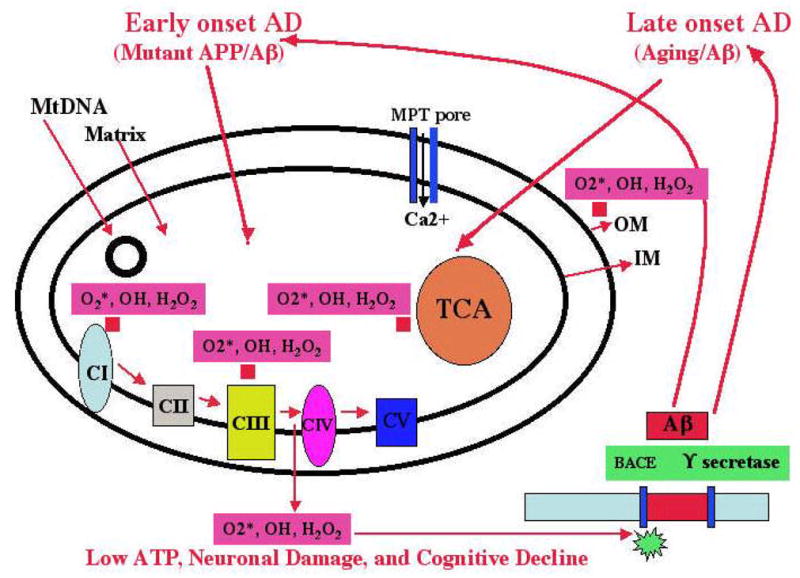
Recent studies of postmortem brains from Alzheimer's disease (AD) patients and transgenic mouse models of AD suggest that oxidative damage, induced by amyloid beta (Abeta), is associated with mitochondria early in AD progression. Abeta and amyloid-precursor protein are known to localize to mitochondrial membranes, block the transport of nuclear-encoded mitochondrial proteins to mitochondria, interact with mitochondrial proteins, disrupt the electron-transport chain, increase reactive oxygen species production, cause mitochondrial damage and prevent neurons from functioning normally. Furthermore, accumulation of Abeta at synaptic terminals might contribute to synaptic damage and cognitive decline in patients with AD. Here, we describe recent studies regarding the roles of Abeta and mitochondrial function in AD progression and particularly in synaptic damage and cognitive decline.
Figures
References
-
- Manczak M, et al. Mitochondria are a direct site of Abeta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. - PubMed
-
- Caspersen C, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–1. - PubMed
-
- Nunomura A, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. - PubMed
-
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
