

Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells
- PMID: 18218921
- PMCID: PMC2493079
- DOI: 10.1096/fj.07-087627
Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells
Abstract
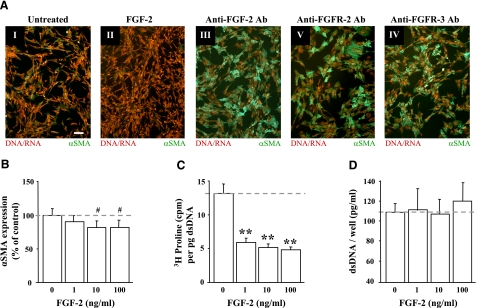
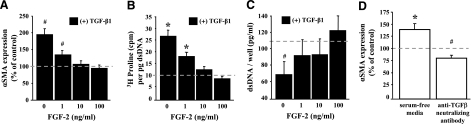
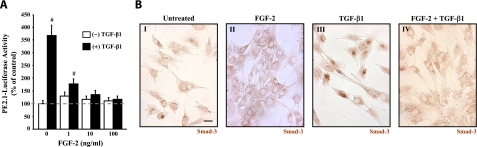
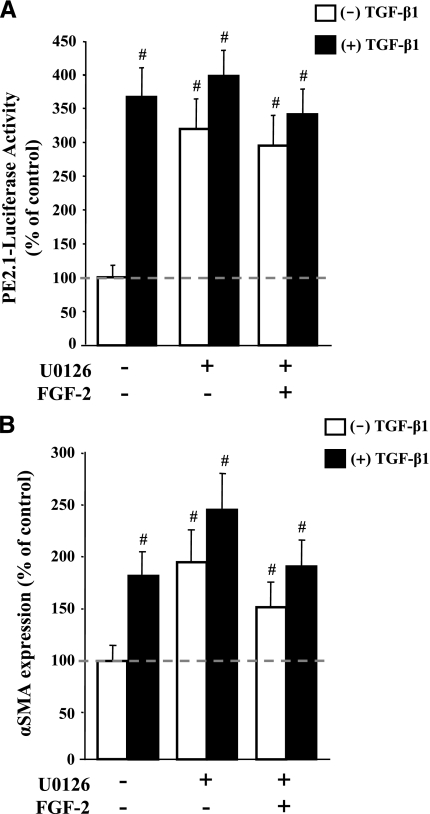
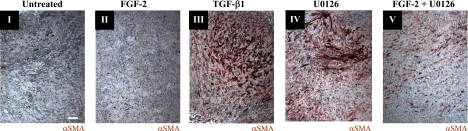
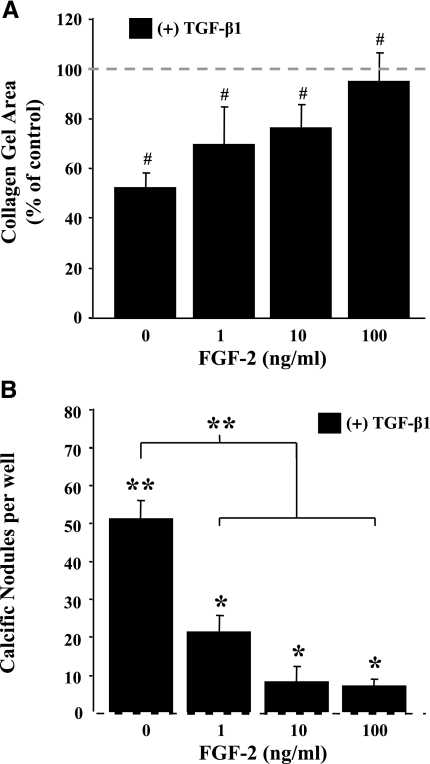
This study aimed to identify signaling pathways that oppose connective tissue fibrosis in the aortic valve. Using valvular interstitial cells (VICs) isolated from porcine aortic valve leaflets, we show that basic fibroblast growth factor (FGF-2) effectively blocks transforming growth factor-beta1 (TGF-beta1)-mediated myofibroblast activation. FGF-2 prevents the induction of alpha-smooth muscle actin (alphaSMA) expression and the exit of VICs from the cell cycle, both of which are hallmarks of myofibroblast activation. By blocking the activity of the Smad transcription factors that serve as the downstream nuclear effectors of TGF-beta1, FGF-2 treatment inhibits fibrosis in VICs. Using an exogenous Smad-responsive transcriptional promoter reporter, we show that Smad activity is repressed by FGF-2, likely an effect of the fact that FGF-2 treatment prevents the nuclear localization of Smads in these cells. This appears to be a direct effect of FGF signaling through mitogen-activated protein kinase (MAPK) cascades as the treatment of VICs with the MAPK/extracellular regulated kinase (MEK) inhibitor U0126 acted to induce fibrosis and blocked the ability of FGF-2 to inhibit TGF-beta1 signaling. Furthermore, FGF-2 treatment of VICs blocks the development of pathological contractile and calcifying phenotypes, suggesting that these pathways may be utilized in the engineering of effective treatments for valvular disease.
Figures
References
-
- Walker G. A., Masters K. S., Shah D. N., Anseth K. S., Leinwand L. A. Valvular myofibroblast activation by transforming growth factor-beta. Circ Res. 2004;95:253–260. - PubMed
-
- Kaden J. J., Kilic R., Sarikoc A., Hagl S., Lang S., Hoffmann U., Brueckmann M., Borggrefe M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med. 2005;16:869–872. - PubMed
-
- Jian B., Narula N., Li Q. Y., Mohler E. R., 3rd, Levy R. J. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. - PubMed
-
- Rabkin-Aikawa E., Farber M., Aikawa M., Schoen F. J. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13:841–847. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
