

The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity
- PMID: 18220271
- PMCID: PMC3025414
- DOI: 10.1002/hep.22183
The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity
Abstract
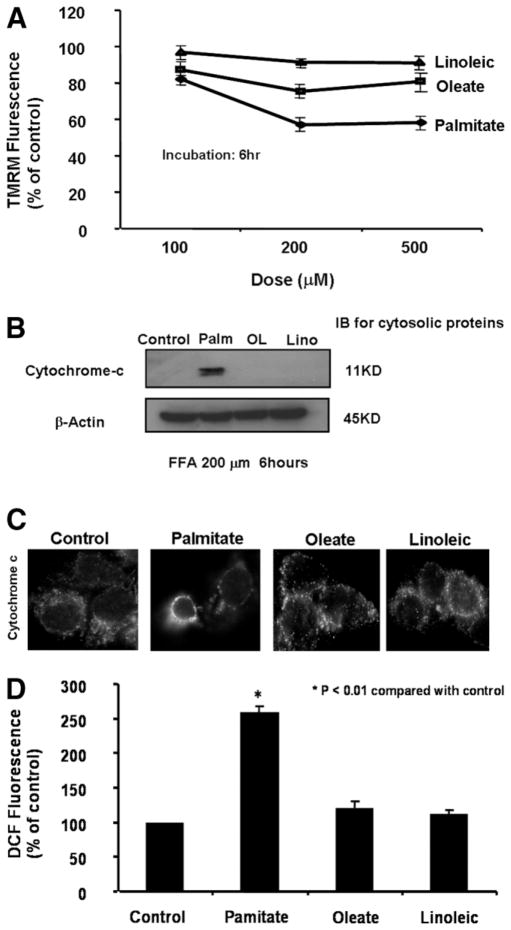
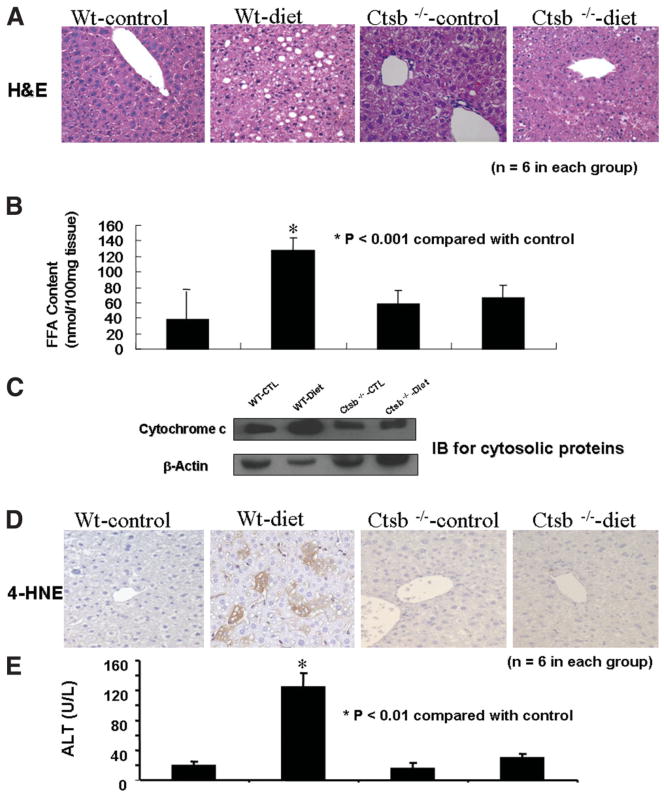
Impaired mitochondrial function is largely thought to be a core abnormality responsible for disease progression in nonalcoholic fatty liver disease (NAFLD). However, the molecular mechanisms resulting in mitochondrial dysfunction in NAFLD remain poorly understood. This study examined the effects of excessive accumulation of free fatty acids (FFAs) in liver cells on mitochondrial function and the role of the lysosomal-mitochondrial axis on lipotoxicity. Primary mouse hepatocytes, HepG2 and McNtcp.24 cells, were treated with varied concentrations of FFAs with different degrees of saturation for up to 24 hours. Mitochondrial function was monitored by real-time imaging, cytochrome c redistribution, and reactive oxygen species (ROS) production. The temporal relationship of lysosomal and mitochondrial permeabilization was established. Activity of the lysosomal protease cathepsin B was suppressed by genetic and pharmacological approaches. Cathepsin B-knockout mice and wild-type animals were place on a high-carbohydrate diet for 16 weeks, and mitochondrial function and liver damage were assessed. Exposure of liver cells to saturated FFAs resulted in mitochondrial depolarization, cytochrome c release, and increased ROS production. Lysosomal permeabilization and cathepsin B redistribution into the cytoplasm occurred several hours prior to mitochondrial dysfunction. Either pharmacological or genetic inhibition of cathepsin B preserved mitochondrial function. Finally, cathepsin B inactivation protected mitochondria, decreased oxidative stress, and attenuated hepatic injury in vivo.
Conclusion: These data strongly suggest excessive accumulation of saturated FFAs in liver cells directly induce mitochondrial dysfunction and oxidative stress. Our data further suggest this process is dependent on lysosomal disruption and activation of cathepsin B.
Figures





References
-
- Wieckowska A, Feldstein AE. Nonalcoholic fatty liver disease in the pediatric population: a review. Curr Opin Pediatr. 2005;17:636–641. - PubMed
-
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. - PubMed
-
- Brunt EM, Neuschwander-Tetri BA, Oliver D, Wehmeier KR, Bacon BR. Nonalcoholic steatohepatitis: histologic features and clinical correlations with 30 blinded biopsy specimens. Hum Pathol. 2004;35:1070–1082. - PubMed
-
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. - PubMed
-
- Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical