

Aggresome formation and neurodegenerative diseases: therapeutic implications
- PMID: 18220762
- PMCID: PMC4403008
- DOI: 10.2174/092986708783330692
Aggresome formation and neurodegenerative diseases: therapeutic implications
Abstract
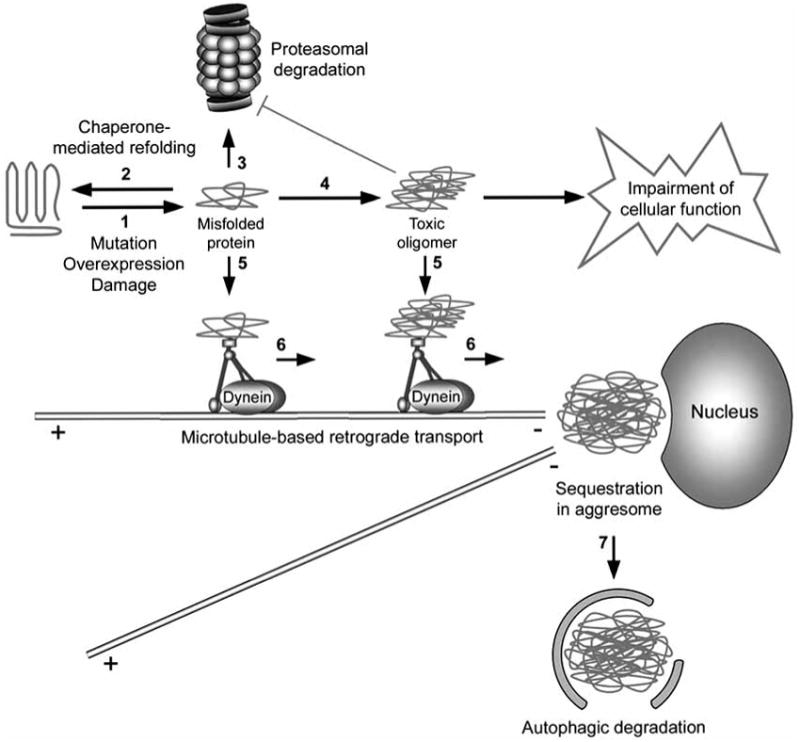
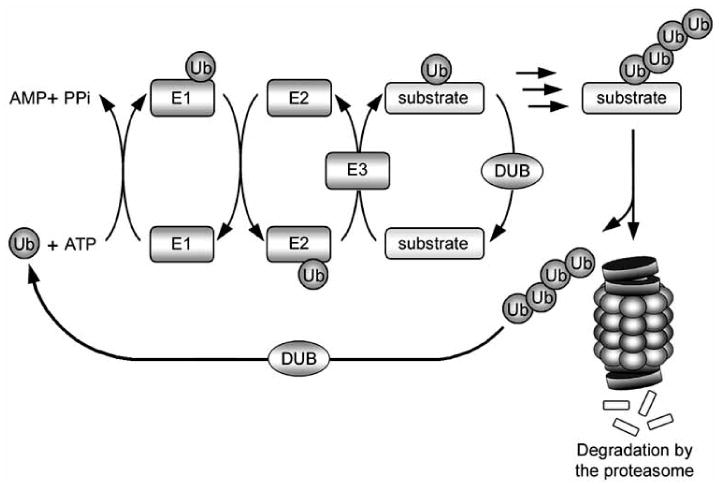
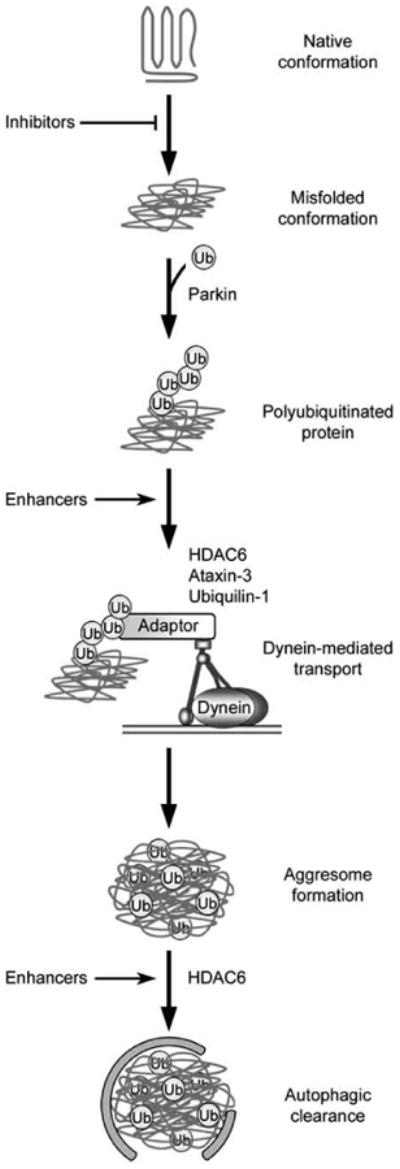
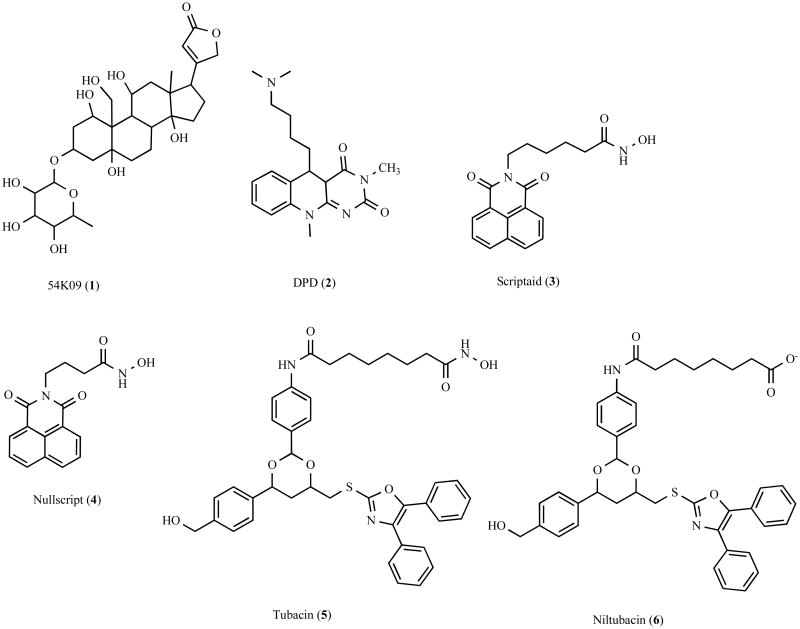
Accumulation of misfolded proteins in proteinaceous inclusions is a prominent pathological feature common to many age-related neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, Huntington's disease, and amyotrophic lateral sclerosis. In cultured cells, when the production of misfolded proteins exceeds the capacity of the chaperone refolding system and the ubiquitin-proteasome degradation pathway, misfolded proteins are actively transported to a cytoplasmic juxtanuclear structure called an aggresome. Aggresome formation is recognized as a cytoprotective response serving to sequester potentially toxic misfolded proteins and facilitate their clearance by autophagy. Recent evidence indicates that aggresome formation is mediated by dynein/dynactin-mediated microtubule-based transport of misfolded proteins to the centrosome and involves several regulators, including histone deacetylase 6, E3 ubiquitin-protein ligase parkin, deubiquitinating enzyme ataxin-3, and ubiquilin-1. Characterization of the molecular mechanisms underlying aggresome formation and its regulation has begun to provide promising therapeutic targets that may be relevant to neurodegenerative diseases. In this review, we provide an overview of the molecular machinery controlling aggresome formation and discuss potential useful compounds and intervention strategies for preventing or reducing the cytotoxicity of misfolded and aggregated proteins.
Figures
References
-
- Ross CA, Poirier MA. Nat Med. 2004;10:S10–17. - PubMed
-
- Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. - PubMed
-
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Nature. 2002;416:507–511. - PubMed
-
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. - PubMed
-
- Fandrich M, Fletcher MA, Dobson CM. Nature. 2001;410:165–166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
