

Dual role of proapoptotic BAD in insulin secretion and beta cell survival
- PMID: 18223655
- PMCID: PMC3918232
- DOI: 10.1038/nm1717
Dual role of proapoptotic BAD in insulin secretion and beta cell survival
Abstract
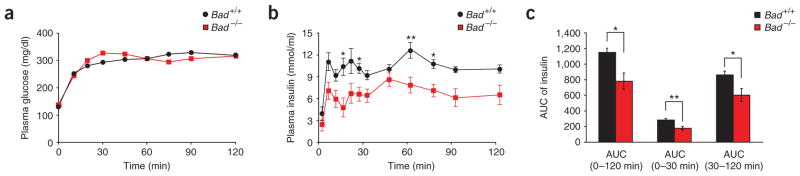
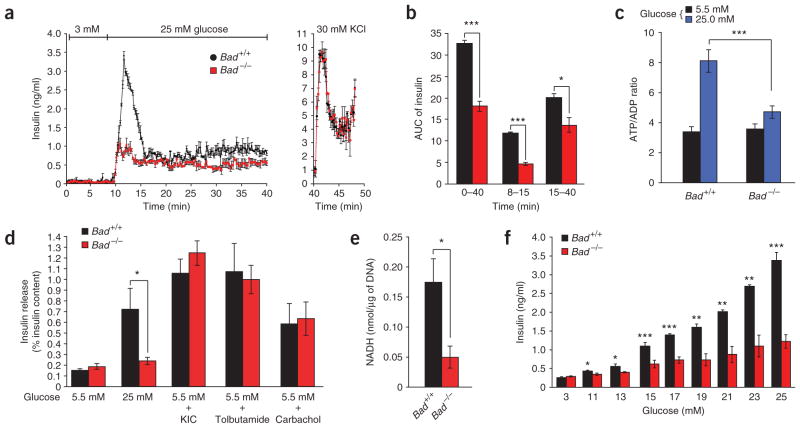
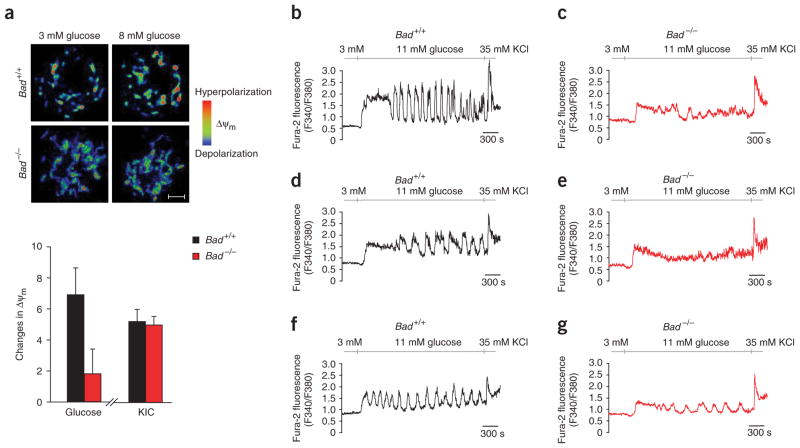
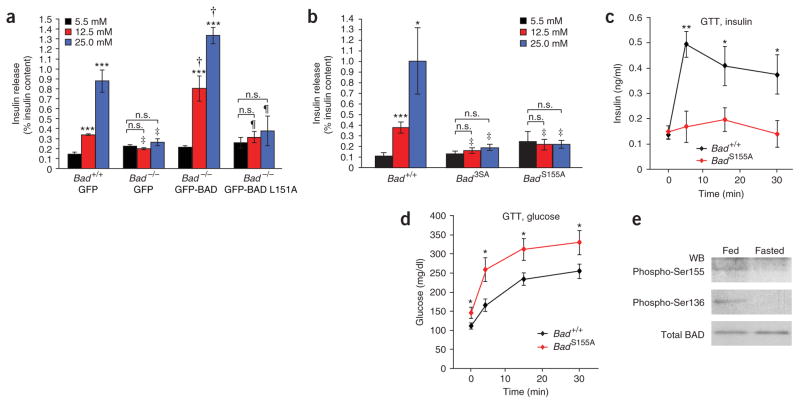
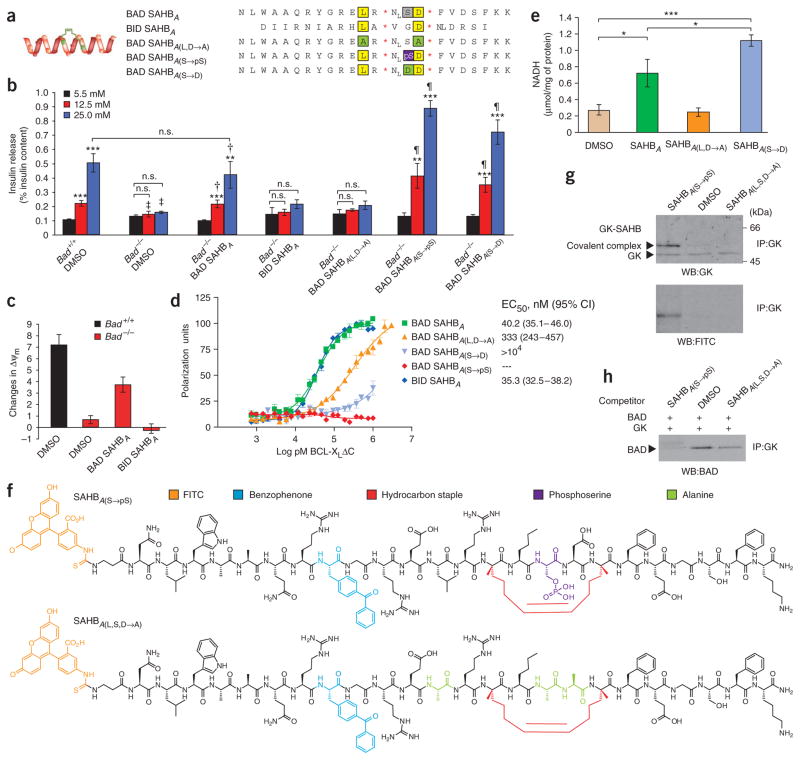
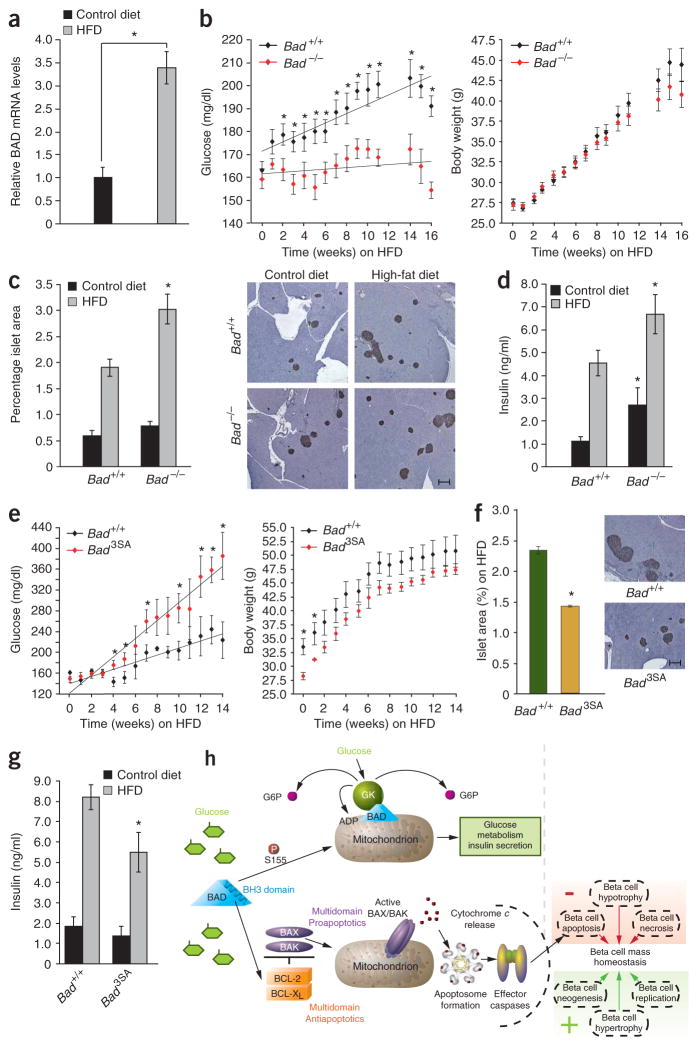
The proapoptotic BCL-2 family member BAD resides in a glucokinase-containing complex that regulates glucose-driven mitochondrial respiration. Here, we present genetic evidence of a physiologic role for BAD in glucose-stimulated insulin secretion by beta cells. This novel function of BAD is specifically dependent upon the phosphorylation of its BH3 sequence, previously defined as an essential death domain. We highlight the pharmacologic relevance of phosphorylated BAD BH3 by using cell-permeable, hydrocarbon-stapled BAD BH3 helices that target glucokinase, restore glucose-driven mitochondrial respiration and correct the insulin secretory response in Bad-deficient islets. Our studies uncover an alternative target and function for the BAD BH3 domain and emphasize the therapeutic potential of phosphorylated BAD BH3 mimetics in selectively restoring beta cell function. Furthermore, we show that BAD regulates the physiologic adaptation of beta cell mass during high-fat feeding. Our findings provide genetic proof of the bifunctional activities of BAD in both beta cell survival and insulin secretion.
Figures
References
-
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. - PubMed
-
- Danial NN, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. - PubMed
-
- Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 2002;13:75–78. - PubMed
-
- Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. - PubMed
-
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
