

Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy
- PMID: 18226499
- PMCID: PMC2272535
- DOI: 10.1016/j.eplepsyres.2007.11.011
Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy
Abstract
Acute brain insults, such as traumatic brain injury, status epilepticus, or stroke are common etiologies for the development of epilepsy, including temporal lobe epilepsy (TLE), which is often refractory to drug therapy. The mechanisms by which a brain injury can lead to epilepsy are poorly understood. It is well recognized that excessive glutamatergic activity plays a major role in the initial pathological and pathophysiological damage. This initial damage is followed by a latent period, during which there is no seizure activity, yet a number of pathophysiological and structural alterations are taking place in key brain regions, that culminate in the expression of epilepsy. The process by which affected/injured neurons that have survived the acute insult, along with well-preserved neurons are progressively forming hyperexcitable, epileptic neuronal networks has been termed epileptogenesis. Understanding the mechanisms of epileptogenesis is crucial for the development of therapeutic interventions that will prevent the manifestation of epilepsy after a brain injury, or reduce its severity. The amygdala, a temporal lobe structure that is most well known for its central role in emotional behavior, also plays a key role in epileptogenesis and epilepsy. In this article, we review the current knowledge on the pathology of the amygdala associated with epileptogenesis and/or epilepsy in TLE patients, and in animal models of TLE. In addition, because a derangement in the balance between glutamatergic and GABAergic synaptic transmission is a salient feature of hyperexcitable, epileptic neuronal circuits, we also review the information available on the role of the glutamatergic and GABAergic systems in epileptogenesis and epilepsy in the amygdala.
Figures
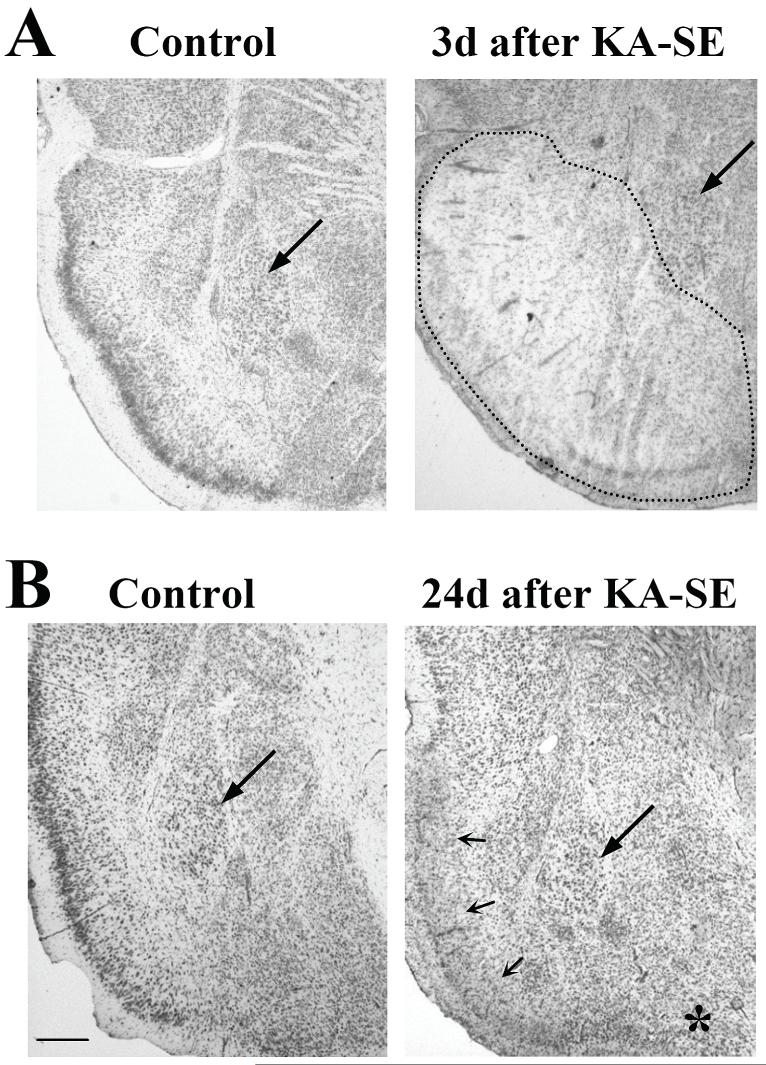
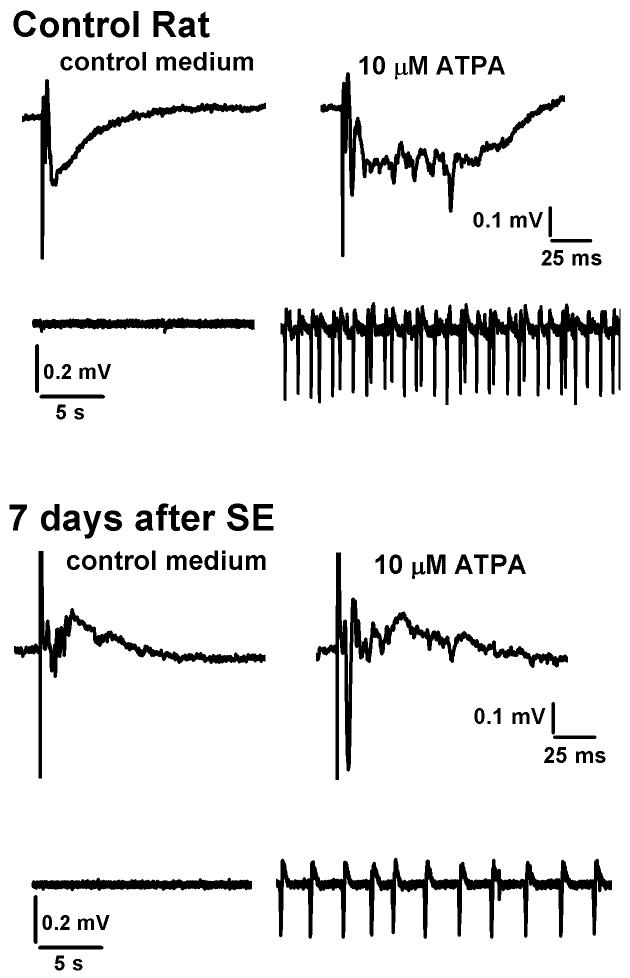
References
-
- Anderson V, Hauser W, Rich S. Genetic heterogeneity and epidemiology of the epilepsies. In: Delgado-Escueta A, Porter R, editors. Jasper’s Basic Mechanisms of the Epilepsies. Lippincott Williams and Wilkins; Philadelphia: 1999. pp. 59–73.
-
- Andre V, Ferrandon A, Marescaux C, Nehlig A. Vigabatrin protects against hippocampal damage but is not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy. Epilepsy Res. 2001;47:99–117. - PubMed
-
- Angeleri F, Majkowski J, Cacchio G, Sobieszek A, D’Acunto S, Gesuita R, Bachleda A, Polonara G, Krolicki L, Signorino M, Salvolini U. Posttraumatic epilepsy risk factors: one-year prospective study after head injury. Epilepsia. 1999;40:1222–1230. - PubMed
-
- Annegers J. The epidemiology of epilepsy. In: Wyllie E, editor. The Treatment of Epilepsy. Lea & Febiger; Philadelphia: 1993. pp. 157–164.
-
- Aroniadou-Anderjaska V, Qashu F, Braga MFM. Mechanisms Regulating GABAergic Inhibitory Transmission in the Basolateral Amygdala: Implications for Epilepsy and Anxiety Disorders. Amino Acids. 2007;32:305–315. - PubMed
