

Validation of protein models by a neural network approach
- PMID: 18230168
- PMCID: PMC2276493
- DOI: 10.1186/1471-2105-9-66
Validation of protein models by a neural network approach
Abstract
Background: The development and improvement of reliable computational methods designed to evaluate the quality of protein models is relevant in the context of protein structure refinement, which has been recently identified as one of the bottlenecks limiting the quality and usefulness of protein structure prediction.
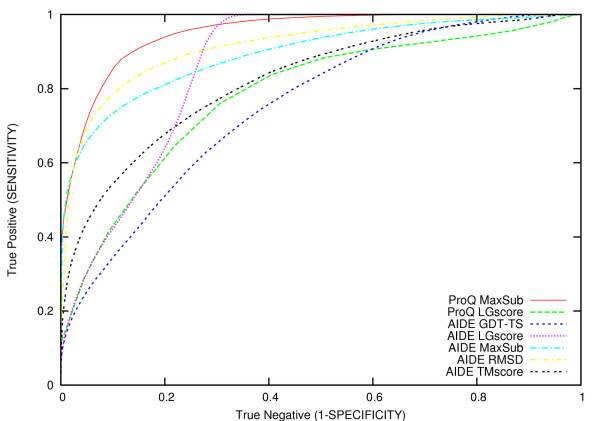
Results: In this contribution, we present a computational method (Artificial Intelligence Decoys Evaluator: AIDE) which is able to consistently discriminate between correct and incorrect protein models. In particular, the method is based on neural networks that use as input 15 structural parameters, which include energy, solvent accessible surface, hydrophobic contacts and secondary structure content. The results obtained with AIDE on a set of decoy structures were evaluated using statistical indicators such as Pearson correlation coefficients, Znat, fraction enrichment, as well as ROC plots. It turned out that AIDE performances are comparable and often complementary to available state-of-the-art learning-based methods.
Conclusion: In light of the results obtained with AIDE, as well as its comparison with available learning-based methods, it can be concluded that AIDE can be successfully used to evaluate the quality of protein structures. The use of AIDE in combination with other evaluation tools is expected to further enhance protein refinement efforts.
Figures
Similar articles
-
Using neural networks and evolutionary information in decoy discrimination for protein tertiary structure prediction.BMC Bioinformatics. 2008 Feb 11;9:94. doi: 10.1186/1471-2105-9-94. BMC Bioinformatics. 2008. PMID: 18267018 Free PMC article.
-
Analysis and identification of beta-turn types using multinomial logistic regression and artificial neural network.Bioinformatics. 2007 Dec 1;23(23):3125-30. doi: 10.1093/bioinformatics/btm324. Epub 2007 Jun 28. Bioinformatics. 2007. Retraction in: Bioinformatics. 2019 Jun 1;35(12):e8-e15. doi: 10.1093/bioinformatics/btm094. PMID: 17599929 Retracted.
-
Artificial neural network models for prediction of intestinal permeability of oligopeptides.BMC Bioinformatics. 2007 Jul 11;8:245. doi: 10.1186/1471-2105-8-245. BMC Bioinformatics. 2007. PMID: 17623108 Free PMC article.
-
OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar.Bioinformatics. 2008 Aug 1;24(15):1662-8. doi: 10.1093/bioinformatics/btn221. Epub 2008 May 12. Bioinformatics. 2008. PMID: 18474507
-
Pattern recognition methods for protein functional site prediction.Curr Protein Pept Sci. 2005 Oct;6(5):479-91. doi: 10.2174/138920305774329322. Curr Protein Pept Sci. 2005. PMID: 16248799 Review.
Cited by
-
Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca²⁺.Front Genet. 2015 May 19;6:185. doi: 10.3389/fgene.2015.00185. eCollection 2015. Front Genet. 2015. PMID: 26136767 Free PMC article.
-
SInCRe-structural interactome computational resource for Mycobacterium tuberculosis.Database (Oxford). 2015 Jun 30;2015:bav060. doi: 10.1093/database/bav060. Print 2015. Database (Oxford). 2015. PMID: 26130660 Free PMC article.
-
A database for human Y chromosome protein data.Bioinformation. 2009 Oct 24;4(5):184-6. doi: 10.6026/97320630004184. Bioinformation. 2009. PMID: 20461156 Free PMC article.
-
The kiwifruit emerging pathogen Pseudomonas syringae pv. actinidiae does not produce AHLs but possesses three luxR solos.PLoS One. 2014 Jan 31;9(1):e87862. doi: 10.1371/journal.pone.0087862. eCollection 2014. PLoS One. 2014. PMID: 24498215 Free PMC article.
-
Structural annotation of Mycobacterium tuberculosis proteome.PLoS One. 2011;6(10):e27044. doi: 10.1371/journal.pone.0027044. Epub 2011 Oct 31. PLoS One. 2011. PMID: 22073123 Free PMC article.
References
-
- Tress M, Ezkurdia I, Grana O, Lopez G, A V. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins: Structure, Function, and Bioinformatics. 2005;61:27–45. - PubMed
-
- Bradley P, Malmstrom L, Qian B, Schonbrun J, Chivian D, Kim D, Meiler J, Misura K, D B. Free modeling with Rosetta in CASP6. Proteins: Structure, Function, and Bioinformatics. 2005;61:128–134. - PubMed
-
- Soonming J, Eunae K, Seokmin S, P Y. Ab inition folding of helix bundle proteins using molecular dynamics simulations. JACS. 2003;125:14841–14846. - PubMed
-
- Andrzej Kolinacuteski JMB. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins: Structure, Function, and Bioinformatics. 2005;61:84–90. - PubMed
-
- Moult J, Fidelis K, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP)-round 6. Proteins: Structure, Function, and Bioinformatics. 2005;61:3–7. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
