

Telomere maintenance and human bone marrow failure
- PMID: 18239083
- PMCID: PMC2343587
- DOI: 10.1182/blood-2007-08-019729
Telomere maintenance and human bone marrow failure
Abstract
Acquired and congenital aplastic anemias recently have been linked molecularly and pathophysiologically by abnormal telomere maintenance. Telomeres are repeated nucleotide sequences that cap the ends of chromosomes and protect them from damage. Telomeres are eroded with cell division, but in hematopoietic stem cells, maintenance of their length is mediated by telomerase. Accelerated telomere shortening is virtually universal in dyskeratosis congenita, caused by mutations in genes encoding components of telomerase or telomere-binding protein (TERT, TERC, DKC1, NOP10, or TINF2). About one-third of patients with acquired aplastic anemia also have short telomeres, which in some cases associate with TERT or TERC mutations. These mutations cause low telomerase activity, accelerated telomere shortening, and diminished proliferative capacity of hematopoietic progenitors. As in other genetic diseases, additional environmental, genetic, and epigenetic modifiers must contribute to telomere erosion and ultimately to disease phenotype. Short telomeres also may cause genomic instability and malignant progression in these marrow failure syndromes. Identification of short telomeres has potential clinical implications: it may be useful in dyskeratosis congenita diagnosis, in suggesting mutations in patients with acquired aplastic anemia, and for selection of suitable hematopoietic stem cell family donors for transplantation in telomerase-deficient patients.
Figures
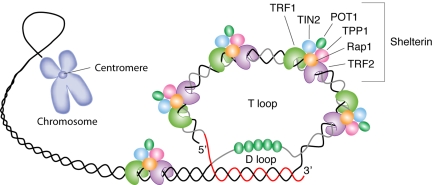
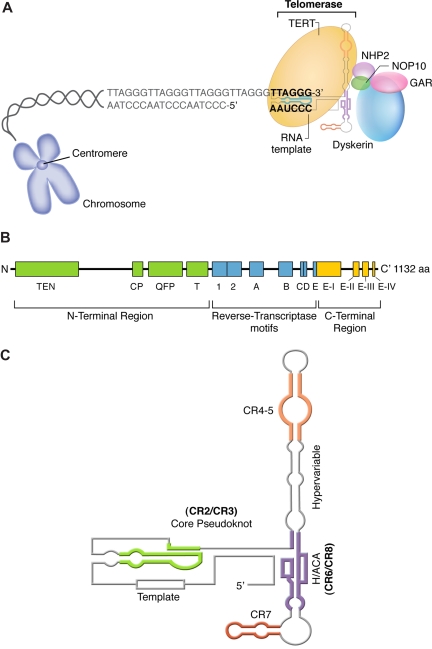
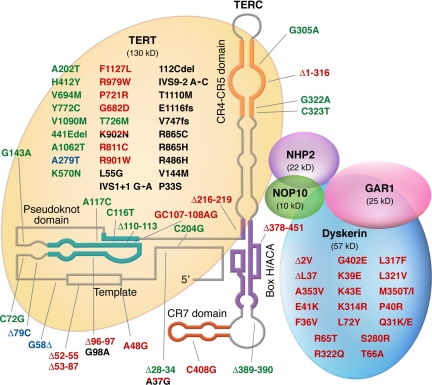
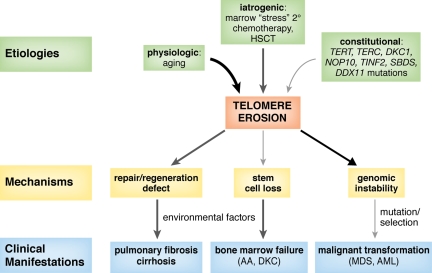
References
-
- Ball SE, Gibson FM, Rizzo S, et al. Progressive telomere shortening in aplastic anemia. Blood. 1998;91:3582–3592. - PubMed
-
- Brummendorf TH, Maciejewski JP, Young NS, Lansdorp PL. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97:895–900. - PubMed
-
- Lee JJ, Kook H, Chung IJ, et al. Telomere length changes in patients with aplastic anaemia. Br J Haematol. 2001;112:1025–1030. - PubMed
-
- Leteurtre F, Li X, Guardiola P, et al. Accelerated telomerase shortening and telomerase activation in Fanconi's anemia. Bone Marrow Transplant. 1999;23:S30.
-
- li X, Leteurtre F, Rocha V, et al. Abnormal telomere metabolism in Fanconi's anaemia correlates with genomic instability and the probability of developing severe aplastic anaemia. Br J Haematol. 2003;120:836–845. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
