

Mitochondrial Ca2+ homeostasis in lysosomal storage diseases
- PMID: 18242695
- PMCID: PMC2517575
- DOI: 10.1016/j.ceca.2007.12.005
Mitochondrial Ca2+ homeostasis in lysosomal storage diseases
Abstract
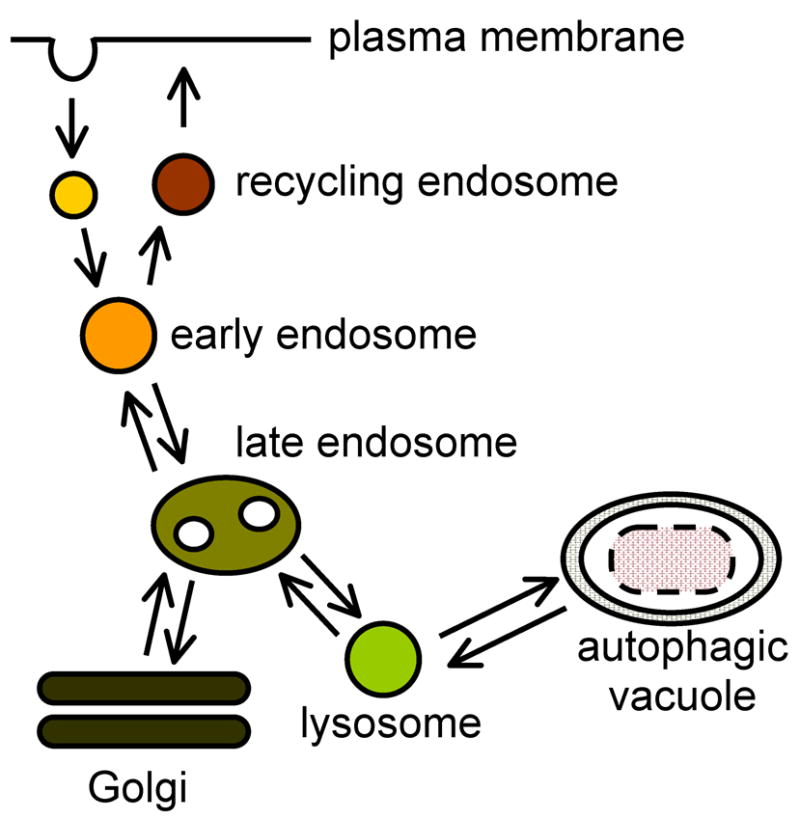
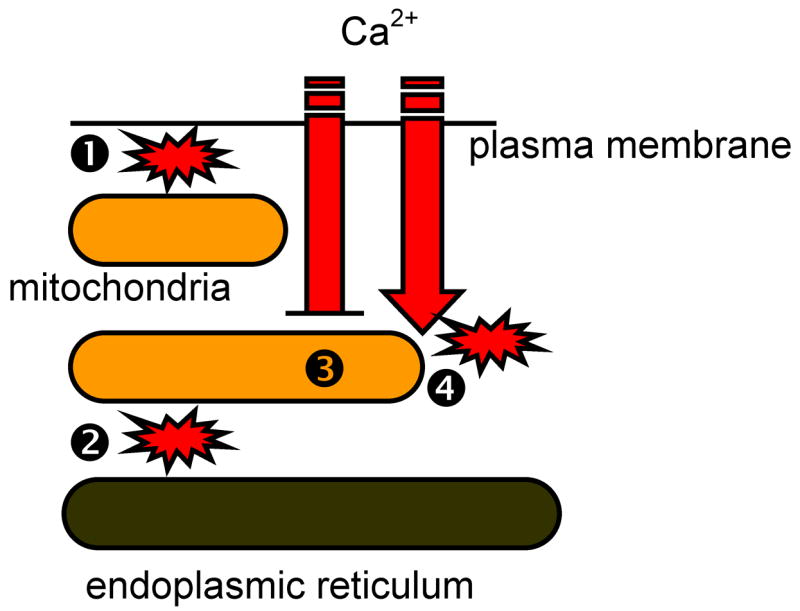
Lysosomal storage diseases (LSDs) are a class of genetic disorders in which proteins responsible for digestion or absorption of endocytosed material do not function or do not localize properly. The resulting cellular "indigestion" causes buildup of intracellular storage inclusions that contain unprocessed lipids and proteins that form macromolecular complexes. The buildup of storage material is associated with degenerative processes that are observed in all LSDs, albeit the correlation between the amount of storage inclusions and the severity of the degenerative processes is not always evident. The latter suggests that a specific mechanism set in motion by aberrant lysosomal function drives the degenerative processes in LSDs. It is becoming increasingly clear that in addition to their function in degrading endocytosed material, lysosomes are essential housekeeping organelles responsible for maintaining healthy population of intracellular organelles, in particular mitochondria. The present review surveys the current knowledge on the lysosomal-mitochondrial axis and its possible role as a contributing factor to mitochondrial Ca(2+) homeostasis and to cell death in LSDs.
Figures


Similar articles
-
Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders.Trends Mol Med. 2017 Feb;23(2):116-134. doi: 10.1016/j.molmed.2016.12.003. Epub 2017 Jan 19. Trends Mol Med. 2017. PMID: 28111024 Review.
-
Lysosomal storage disorders - challenges, concepts and avenues for therapy: beyond rare diseases.J Cell Sci. 2019 Jan 16;132(2):jcs221739. doi: 10.1242/jcs.221739. J Cell Sci. 2019. PMID: 30651381 Review.
-
Lysosomal positioning diseases: beyond substrate storage.Open Biol. 2022 Oct;12(10):220155. doi: 10.1098/rsob.220155. Epub 2022 Oct 26. Open Biol. 2022. PMID: 36285443 Free PMC article. Review.
-
Aberrant Ca2+ handling in lysosomal storage disorders.Cell Calcium. 2010 Feb;47(2):103-11. doi: 10.1016/j.ceca.2009.12.007. Epub 2010 Jan 6. Cell Calcium. 2010. PMID: 20053447 Free PMC article. Review.
-
Autophagy, mitochondria and cell death in lysosomal storage diseases.Autophagy. 2007 May-Jun;3(3):259-62. doi: 10.4161/auto.3906. Epub 2007 May 23. Autophagy. 2007. PMID: 17329960 Free PMC article.
Cited by
-
Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy.Cells. 2021 Mar 5;10(3):572. doi: 10.3390/cells10030572. Cells. 2021. PMID: 33807817 Free PMC article.
-
Effects of chronic exposure to cigarette smoke on canonical transient receptor potential expression in rat pulmonary arterial smooth muscle.Am J Physiol Cell Physiol. 2014 Feb 15;306(4):C364-73. doi: 10.1152/ajpcell.00048.2013. Epub 2013 Dec 11. Am J Physiol Cell Physiol. 2014. PMID: 24336649 Free PMC article.
-
Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA.Mol Genet Metab. 2013 Sep-Oct;110(1-2):54-64. doi: 10.1016/j.ymgme.2013.04.002. Epub 2013 Apr 10. Mol Genet Metab. 2013. PMID: 23665161 Free PMC article. Review.
-
Three-dimensional structure of Rubella virus factories.Virology. 2010 Sep 30;405(2):579-91. doi: 10.1016/j.virol.2010.06.043. Epub 2010 Jul 23. Virology. 2010. PMID: 20655079 Free PMC article.
-
Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts?Hum Mol Genet. 2011 Apr 15;20(R1):R54-60. doi: 10.1093/hmg/ddr112. Epub 2011 Mar 19. Hum Mol Genet. 2011. PMID: 21421999 Free PMC article. Review.
References
-
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–32. - PubMed
-
- Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol Rev. 1997;77:759–803. - PubMed
-
- Bright NA, Reaves BJ, Mullock BM, Luzio JP. Dense core lysosomes can fuse with late endosomes and are re-formed from the resultant hybrid organelles. J Cell Sci. 1997;110(Pt 17):2027–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
