

Prion diseases: from protein to cell pathology
- PMID: 18245809
- PMCID: PMC2258253
- DOI: 10.2353/ajpath.2008.070442
Prion diseases: from protein to cell pathology
Abstract
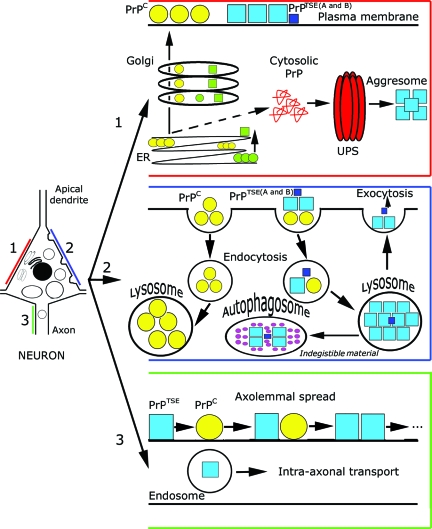
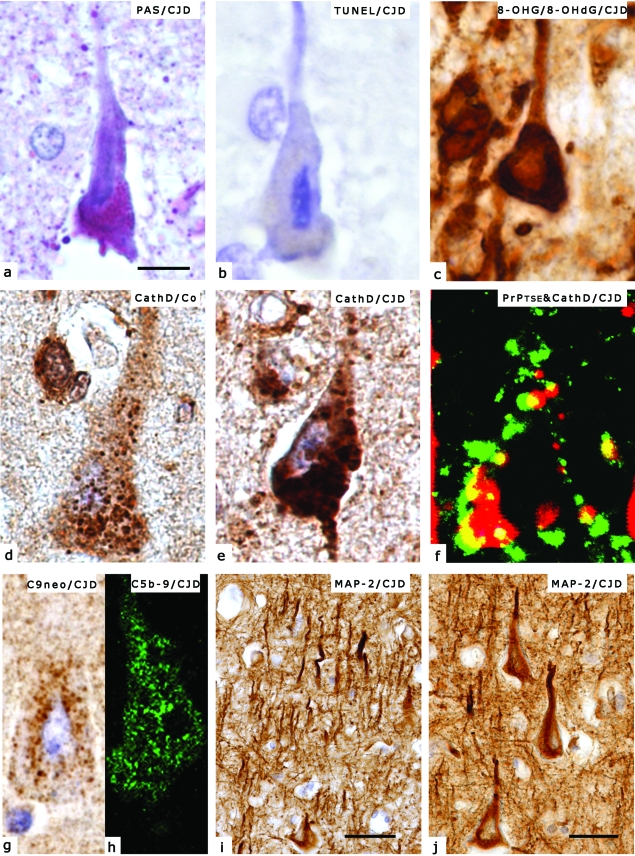
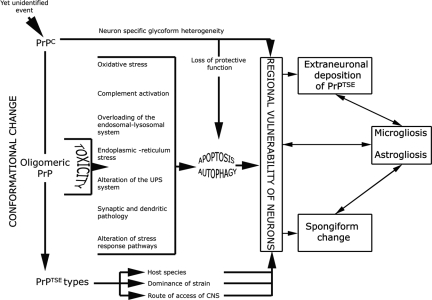
Prion diseases or transmissible spongiform encephalopathies are fatal neurodegenerative conditions in humans and animals that originate spontaneously, genetically or by infection. Conformational change of the normal (cellular) form of prion protein (PrP c) to a pathological, disease-associated form (PrP TSE) is considered central to pathogenesis and formation of the infectious agent or prion. Neuronal damage is central to clinical manifestation of prion diseases but poorly understood. In this review, we analyze the major pathogenetic pathways that lead to tissue pathology in different forms of disease. Neuropathogenesis of prion diseases evolves in complex ways on several front lines, most but not all of which exist also in other neurodegenerative as well as infectious diseases. Whereas intracellular accumulation of PrP forms might significantly impair cell function and lead to cytopathology, mere extracellular deposition of PrP TSE is questionable as a direct cytotoxic factor. Tissue damage may result from several parallel, interacting, or subsequent pathways. Future studies should clarify the trigger(s) and sequence of these processes and whether, and which, one is dominating or decisive.
Figures
References
-
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol. 2006;4:765–775. - PubMed
-
- Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. - PubMed
-
- Ironside JW, Ritchie DL, Head MW. Phenotypic variability in human prion diseases. Neuropathol Appl Neurobiol. 2005;31:565–579. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
