

Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function
- PMID: 18246078
- PMCID: PMC3093709
- DOI: 10.1038/nm1725
Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function
Abstract
Treacher Collins syndrome (TCS) is a congenital disorder of craniofacial development arising from mutations in TCOF1, which encodes the nucleolar phosphoprotein Treacle. Haploinsufficiency of Tcof1 perturbs mature ribosome biogenesis, resulting in stabilization of p53 and the cyclin G1-mediated cell-cycle arrest that underpins the specificity of neuroepithelial apoptosis and neural crest cell hypoplasia characteristic of TCS. Here we show that inhibition of p53 prevents cyclin G1-driven apoptotic elimination of neural crest cells while rescuing the craniofacial abnormalities associated with mutations in Tcof1 and extending life span. These improvements, however, occur independently of the effects on ribosome biogenesis; thus suggesting that it is p53-dependent neuroepithelial apoptosis that is the primary mechanism underlying the pathogenesis of TCS. Our work further implies that neuroepithelial and neural crest cells are particularly sensitive to cellular stress during embryogenesis and that suppression of p53 function provides an attractive avenue for possible clinical prevention of TCS craniofacial birth defects and possibly those of other neurocristopathies.
Figures

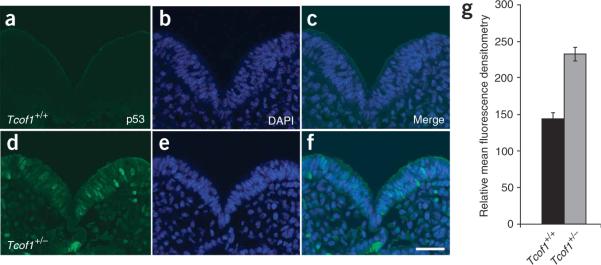

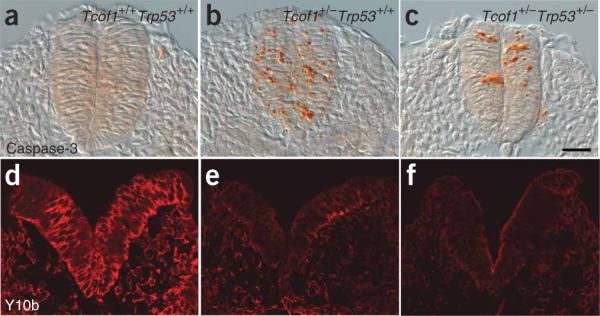
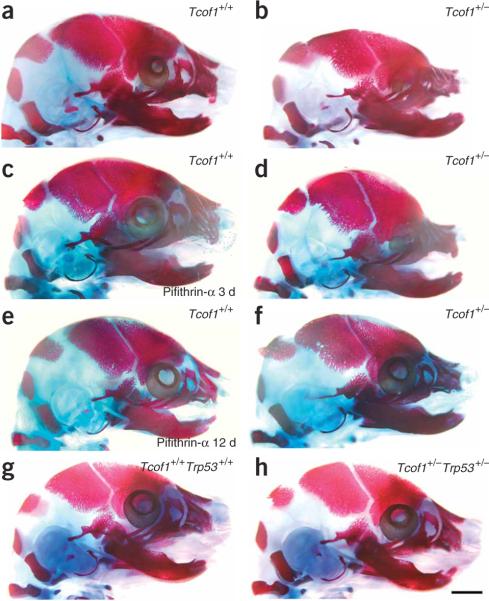
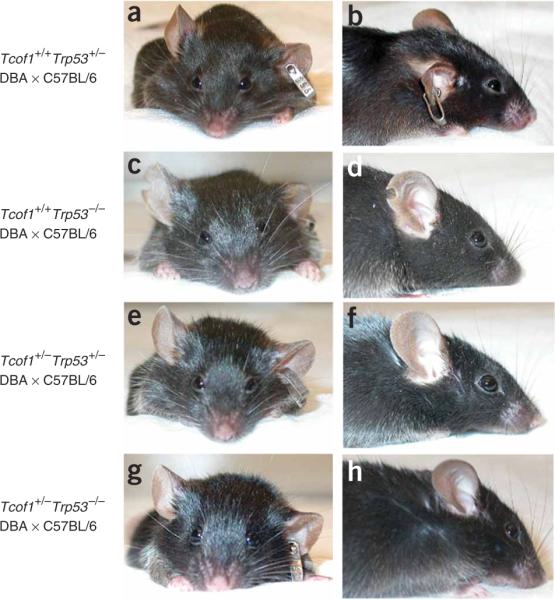
Comment in
-
Saving face: rescuing a craniofacial birth defect.Nat Med. 2008 Feb;14(2):115-6. doi: 10.1038/nm0208-115. Nat Med. 2008. PMID: 18256609 No abstract available.
References
-
- Phelps PD, Poswillo D, Lloyd GA. The ear deformities in mandibulofacial dysostosis (Treacher Collins syndrome) Clin. Otolaryngol. 1981;6:15–28. - PubMed
-
- Rovin S, Dachi SF, Borenstein DB, Cotter WB. Mandibulofacial dysostosis, a familial study of five generations. J. Pediatr. 1964;65:215–221. - PubMed
-
- Fazen LE, Elmore J, Nadler HL. Mandibulo-facial dysostosis. (Treacher-Collins syndrome) Am. J. Dis. Child. 1967;113:405–410. - PubMed
-
- Gladwin AJ, et al. Treacher Collins syndrome may result from insertions, deletions or splicing mutations, which introduce a termination codon into the gene. Hum. Mol. Genet. 1996;5:1533–1538. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
