

Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17
- PMID: 18250191
- PMCID: PMC2271004
- DOI: 10.1084/jem.20071507
Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17
Abstract
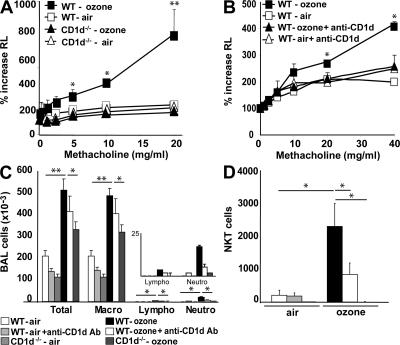
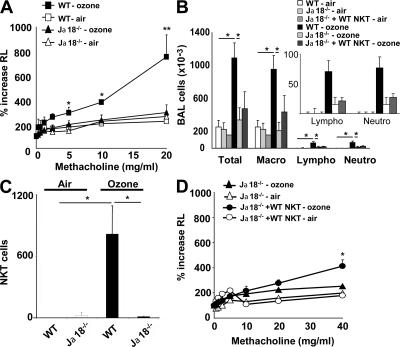
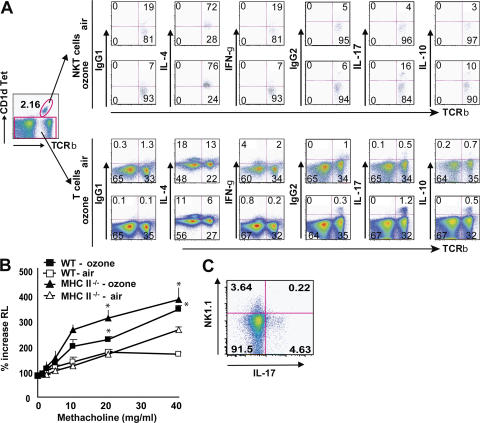
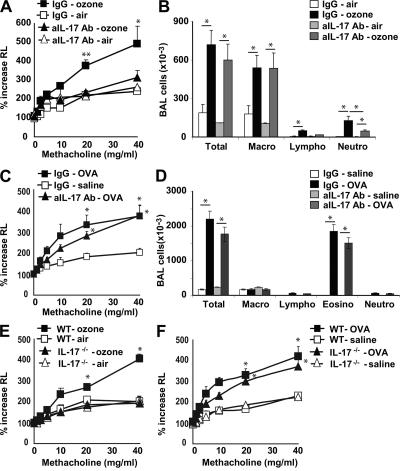
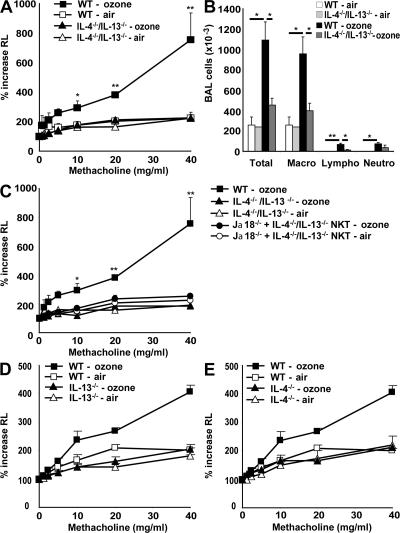
Exposure to ozone, which is a major component of air pollution, induces a form of asthma that occurs in the absence of adaptive immunity. Although ozone-induced asthma is characterized by airway neutrophilia, and not eosinophilia, it is nevertheless associated with airway hyperreactivity (AHR), which is a cardinal feature of asthma. Because AHR induced by allergens requires the presence of natural killer T (NKT) cells, we asked whether ozone-induced AHR had similar requirements. We found that repeated exposure of wild-type (WT) mice to ozone induced severe AHR associated with an increase in airway NKT cells, neutrophils, and macrophages. Surprisingly, NKT cell-deficient (CD1d(-/-) and Jalpha18(-/-)) mice failed to develop ozone-induced AHR. Further, treatment of WT mice with an anti-CD1d mAb blocked NKT cell activation and prevented ozone-induced AHR. Moreover, ozone-induced, but not allergen-induced, AHR was associated with NKT cells producing interleukin (IL)-17, and failed to occur in IL-17(-/-) mice nor in WT mice treated with anti-IL-17 mAb. Thus, ozone exposure induces AHR that requires the presence of NKT cells and IL-17 production. Because NKT cells are required for the development of two very disparate forms of AHR (ozone- and allergen-induced), our results strongly suggest that NKT cells mediate a unifying pathogenic mechanism for several distinct forms of asthma, and represent a unique target for effective asthma therapy.
Figures
References
-
- Gold, D.R., and R. Wright. 2005. Population disparities in asthma. Annu. Rev. Public Health. 26:89–113. - PubMed
-
- Wills-Karp, M. 1999. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 17:255–281. - PubMed
-
- Gibson, P.G., J.L. Simpson, and N. Saltos. 2001. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest. 119:1329–1336. - PubMed
-
- Li, N., J. Alam, M.I. Venkatesan, A. Eiguren-Fernandez, D. Schmitz, E. Di Stefano, N. Slaughter, E. Killeen, X. Wang, A. Huang, et al. 2004. Nrf 2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 173:3467–3481. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
