

Review
doi: 10.1038/nrg2270.
Genome-wide approaches to studying chromatin modifications
Affiliations
- PMID: 18250624
- PMCID: PMC10882563
- DOI: 10.1038/nrg2270
Item in Clipboard
Review
Genome-wide approaches to studying chromatin modifications
Nat Rev Genet.
2008 Mar.
Abstract
Over two metres of DNA is packaged into each nucleus in the human body in a manner that still allows for gene regulation. This remarkable feat is accomplished by the wrapping of DNA around histone proteins in repeating units of nucleosomes to form a structure known as chromatin. This chromatin structure is subject to various modifications that have profound influences on gene expression. Recently developed techniques to study chromatin modifications at a genome-wide scale are now allowing researchers to probe the complex components that make up epigenomes. Here we review genome-wide approaches to studying epigenomic structure and the exciting findings that have been obtained using these technologies.
Figures

a | Restriction enzyme. DNA methylation can be identified using restriction enzymes that differentially recognize methylated and unmethylated cytosine bases. The recognition site for HpaII (CCGG) is shown as an example. Other restriction enzymes (and recognition sites) include AciI (CCGC and GCGG), BstUI (CGCG), HhaI (GCGC) and TaiI (ACGT). b | Bisulphite treatment. The treatment of DNA with bilsulphite changes all unmethylated cytosines to uracils, leaving methylated cytosines unchanged. c | Immunoprecipitation of methylated DNA (mCIP). DNA is first sonicated and an antibody that is specific to methylated cytosines is used to pull down methylated regions. Any of these methods can be combined with either hybridization to DNA microarrays or direct sequencing to study DNA methylation on a genomic scale.

The ChIP–chip method can be used to study many of the epigenomic phenomena discussed in this Review. The example presented here shows how ChIP–chip can be used to study histone modifications. Modified chromatin is first purified by immunoprecipitating crosslinked chromatin using an antibody that is specific to a particular histone modification (shown in green). DNA is then amplified to obtain sufficient DNA. The colour-labelled ChIP DNA, together with the control DNA prepared from input chromatin and labelled with a different colour, is hybridized to a DNA microarray. The microarray probes can then be mapped to the genome to yield genomic coordinates.

The combination of ChIP experiments with SAGE can be used to profile histone modifications at a genomic scale. The ChIP–SAGE procedure begins with a ChIP step to purify chromatin regions that are associated with a specific histone modification (shown in green), and proceeds as follows. First, crosslinks are reversed, a biotinylated universal linker (UL) is ligated to DNA ends and DNA is bound to streptavidin beads. Then NlaIII, which recognizes CATG, is used to digest DNA and a linker containing the recognition sequence of MmeI is ligated to the cleaved DNA ends. MmeI digestion produces 21–22 bp sequence tags from the immunoprecipitated fragments; the sequence tags are concatenated, cloned into a sequencing vector and sequenced. About 20 to 30 short sequence tags of 21 bp can be generated from each sequencing reaction. The sequence tags can then be mapped to the genome to identify modified regions.
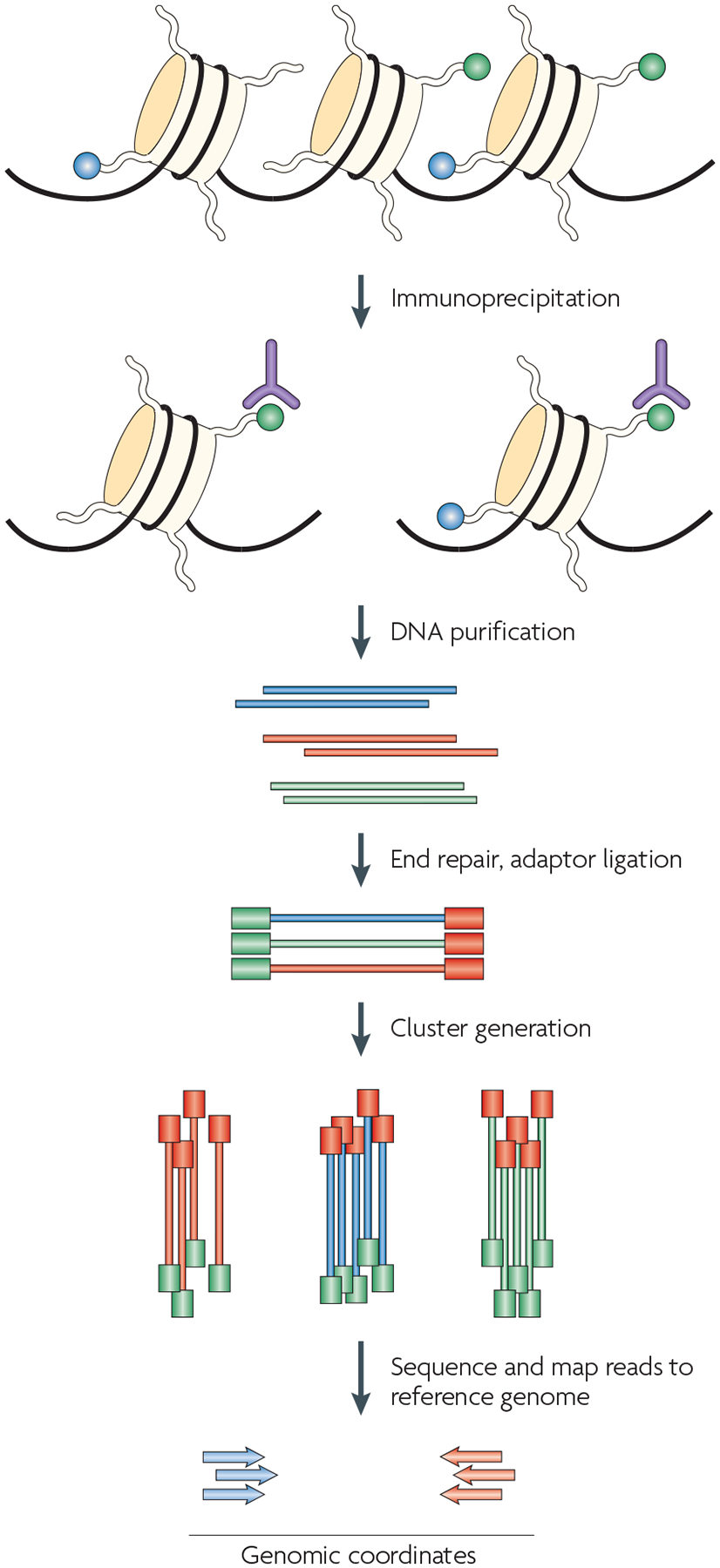
One of the most exciting recent advances in technologies for studying epigenetic phenomena at a genomic scale relies on the combination of ChIP experiments with high-throughput sequencing. The procedure that is outlined here is specific to the Illumina Genome Analyzer using Solexa technology, although other high-throughput sequencing techniques would also work in principle. The first step is the purification of modified chromatin by immunoprecipitation using an antibody that is specific to a particular histone modification (shown in green). The ChIP DNA ends are repaired and ligated to a pair of adaptors, followed by limited PCR amplification. The DNA molecules are bound to the surface of a flow cell that contains covalently bound oligonucleotides that recognize the adaptor sequences. Clusters of individual DNA molecules are generated by solid-phase PCR and sequencing by synthesis is performed. The resulting sequence reads are mapped to a reference genome to obtain genomic coordinates that correspond to the immunoprecipitated fragments.
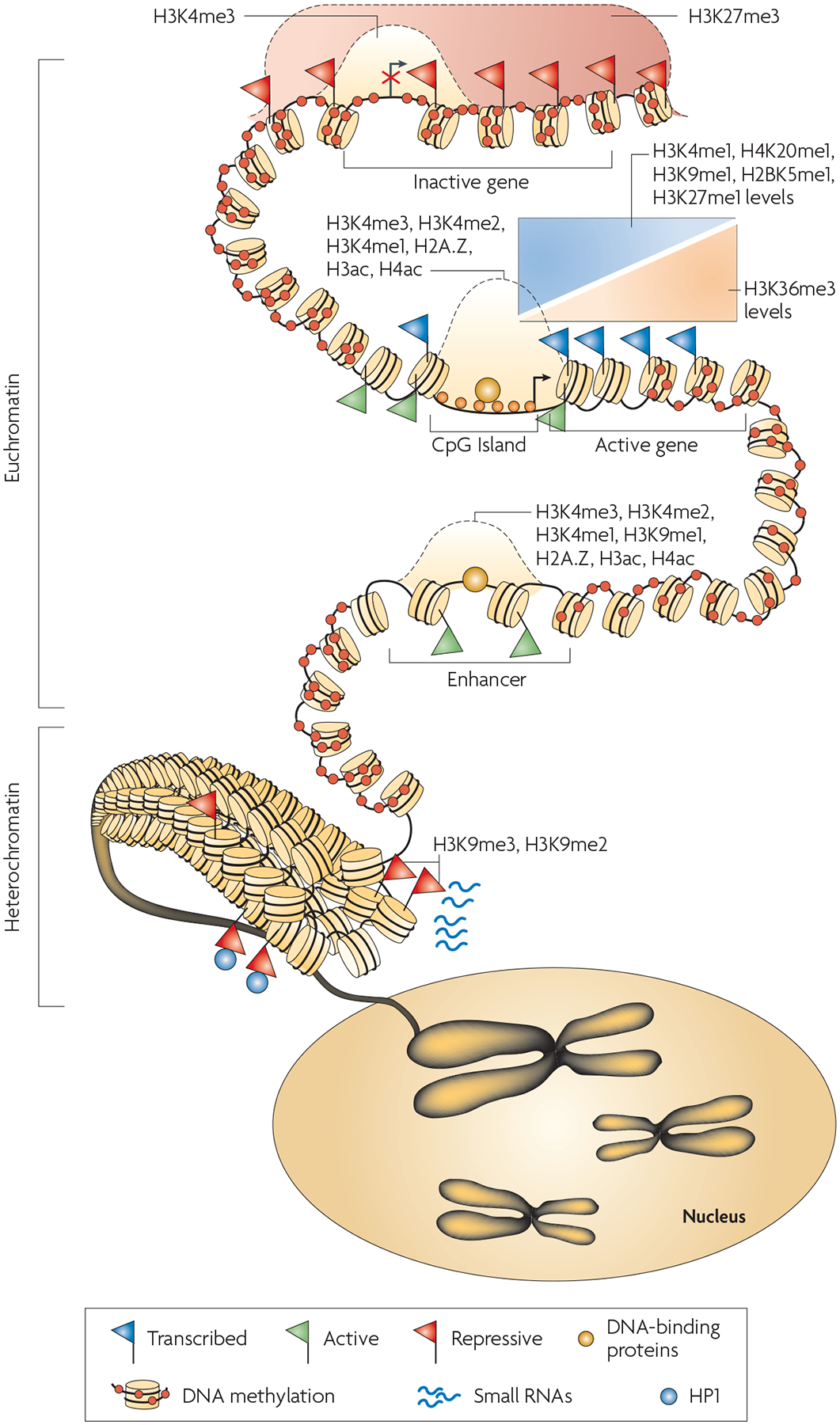
The interaction of DNA methylation, histone modification, nucleosome positioning and other factors such as small RNAs contribute to an overall epigenome that regulates gene expression and allows cells to remember their identity. Chromosomes are divided into accessible regions of euchromatin and poorly accessible regions of heterochromatin. Heterochromatic regions are marked with histone H3 lysine 9 di- and trimethylation (H3K9me2 and H3K9me3), which serve as a platform for HP1 (heterochromatic protein 1) binding. Small RNAs have been implicated in the maintenance of heterochromatin. DNA methylation is persistent throughout genomes, and is missing only in regions such as CpG islands, promoters and possibly enhancers. The H3K27me3 modification is present in broad domains that encompass inactive genes. Histone modifications including H3K4me3, H3K4me2, H3K4me1 as well as histone acetylation and histone variant H2A.Z mark the transcription start site regions of active genes. The monomethylations of H3K4, H3K9, H3K27, H4K20 and H2BK5 mark actively transcribed regions, peaking near the 5′ end of genes. The trimethylation of H3K36 also marks actively transcribed regions, but peaks near the 3′ end of genes.
References
-
- Goldberg AD, Allis CD & Bernstein E Epigenetics: a landscape takes shape. Cell 128, 635–638 (2007). - PubMed
-
- Ptashne M On the use of the word ‘epigenetic’. Curr. Biol 17, R233–R236 (2007). - PubMed
-
- Bird A Perceptions of epigenetics. Nature 447, 396–398 (2007). - PubMed
-
- Bernstein BE, Meissner A & Lander ES The mammalian epigenome. Cell 128, 669–681 (2007). - PubMed
-
- Orlando V & Paro R Mapping Polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell 75, 1187–1198 (1993). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
