

Statistical issues in the analysis of Illumina data
- PMID: 18254947
- PMCID: PMC2291044
- DOI: 10.1186/1471-2105-9-85
Statistical issues in the analysis of Illumina data
Abstract
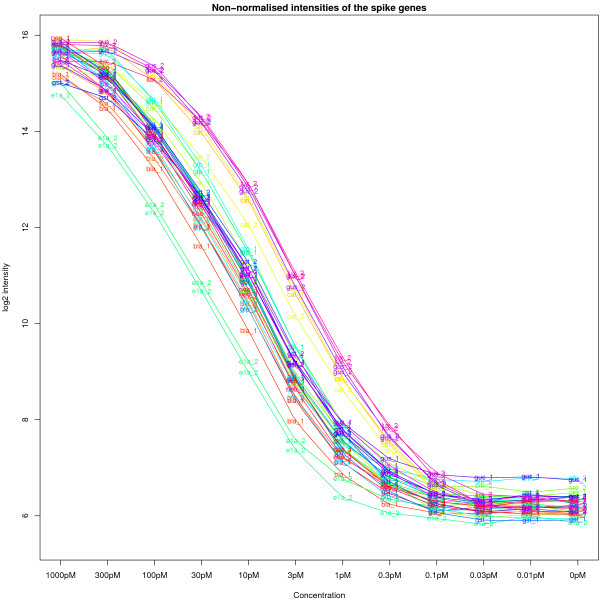
Background: Illumina bead-based arrays are becoming increasingly popular due to their high degree of replication and reported high data quality. However, little attention has been paid to the pre-processing of Illumina data. In this paper, we present our experience of analysing the raw data from an Illumina spike-in experiment and offer guidelines for those wishing to analyse expression data or develop new methodologies for this technology.
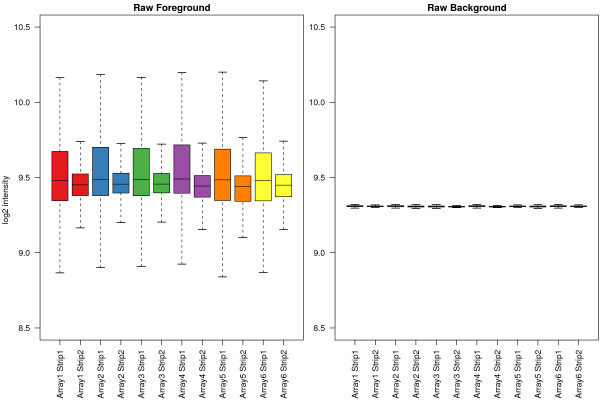
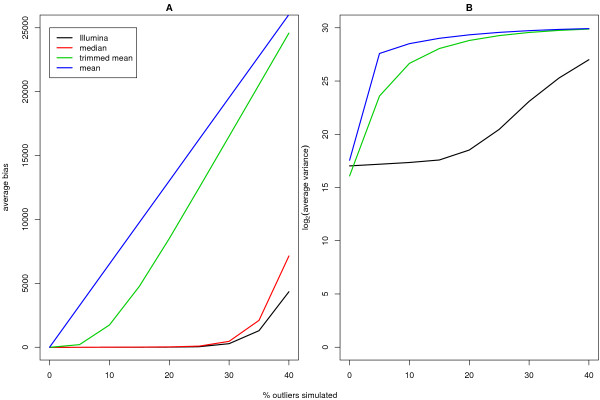
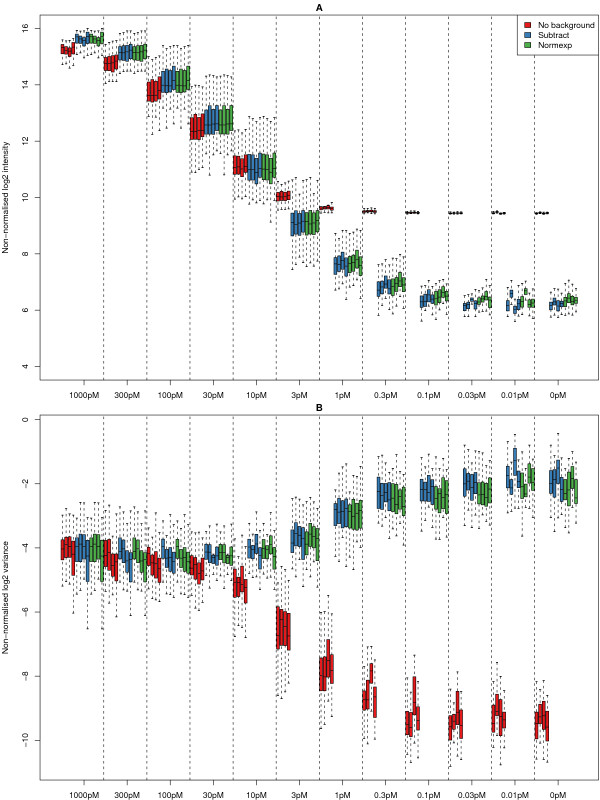
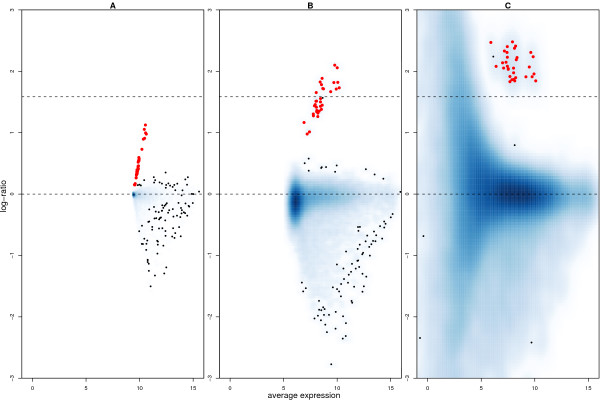
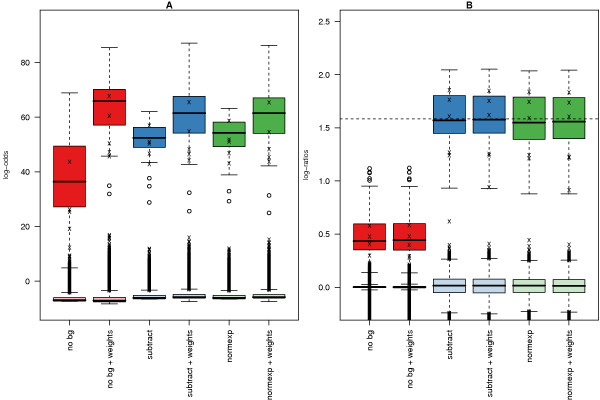
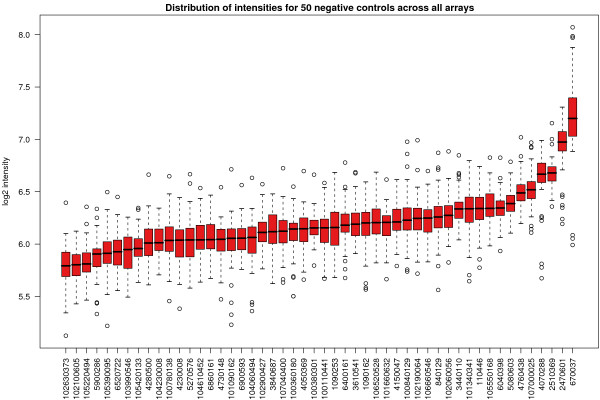
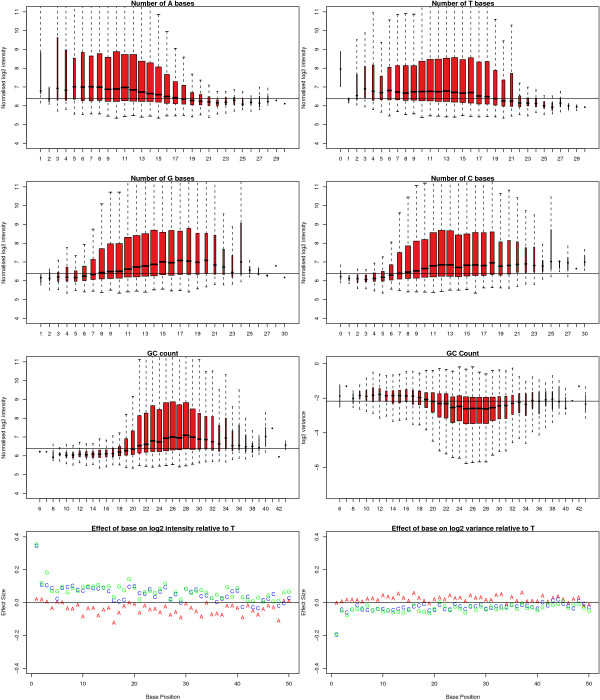
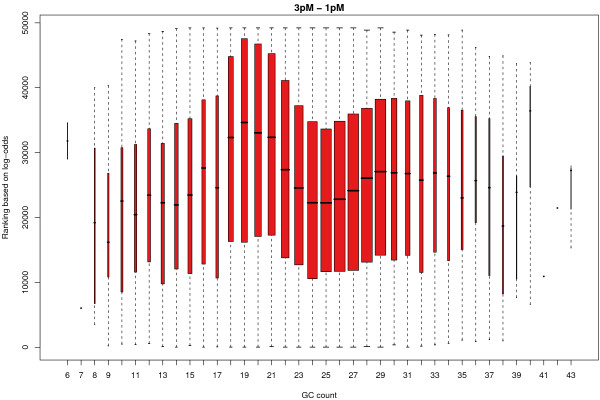
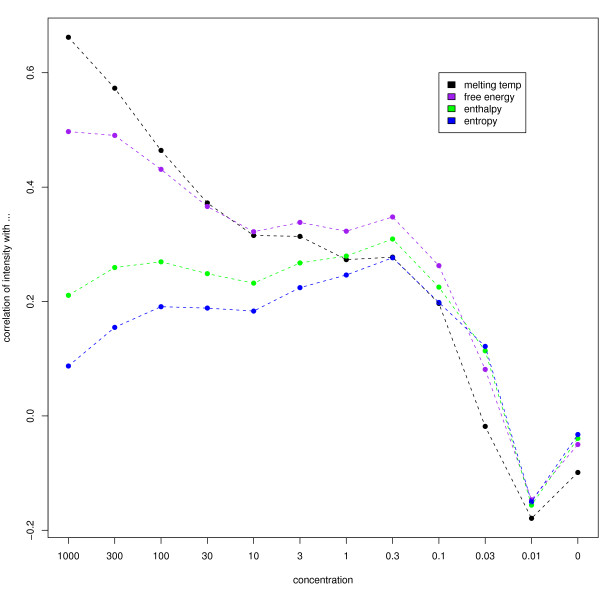
Results: We find that the local background estimated by Illumina is consistently low, and subtracting this background is beneficial for detecting differential expression (DE). Illumina's summary method performs well at removing outliers, producing estimates which are less biased and are less variable than other robust summary methods. However, quality assessment on summarised data may miss spatial artefacts present in the raw data. Also, we find that the background normalisation method used in Illumina's proprietary software (BeadStudio) can cause problems with a standard DE analysis. We demonstrate that variances calculated from the raw data can be used as inverse weights in the DE analysis to improve power. Finally, variability in both expression levels and DE statistics can be attributed to differences in probe composition. These differences are not accounted for by current analysis methods and require further investigation.
Conclusion: Analysing Illumina expression data using BeadStudio is reasonable because of the conservative estimates of summary values produced by the software. Improvements can however be made by not using background normalisation. Access to the raw data allows for a more detailed quality assessment and flexible analyses. In the case of a gene expression study, data can be analysed on an appropriate scale using established tools. Similar improvements can be expected for other Illumina assays.
Figures
References
-
- Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, Doucet D, Milewski M, Yang R, Siegmund C, Haas J, Zhou L, Oliphant A, Fan JB, Barnard S, Chee MS. Decoding randomly ordered DNA arrays. Genome Res. 2004;14:870–877. doi: 10.1101/gr.2255804. - DOI - PMC - PubMed
-
- Dunning MJ, Thorne NP, Camilier I, Smith ML, Tavaré S. Quality control and low-level statistical analysis of Illumina BeadArrays. RevStat. 2006;4:1–30.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
