

Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons
- PMID: 18256268
- PMCID: PMC6671582
- DOI: 10.1523/JNEUROSCI.2723-07.2008
Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons
Abstract
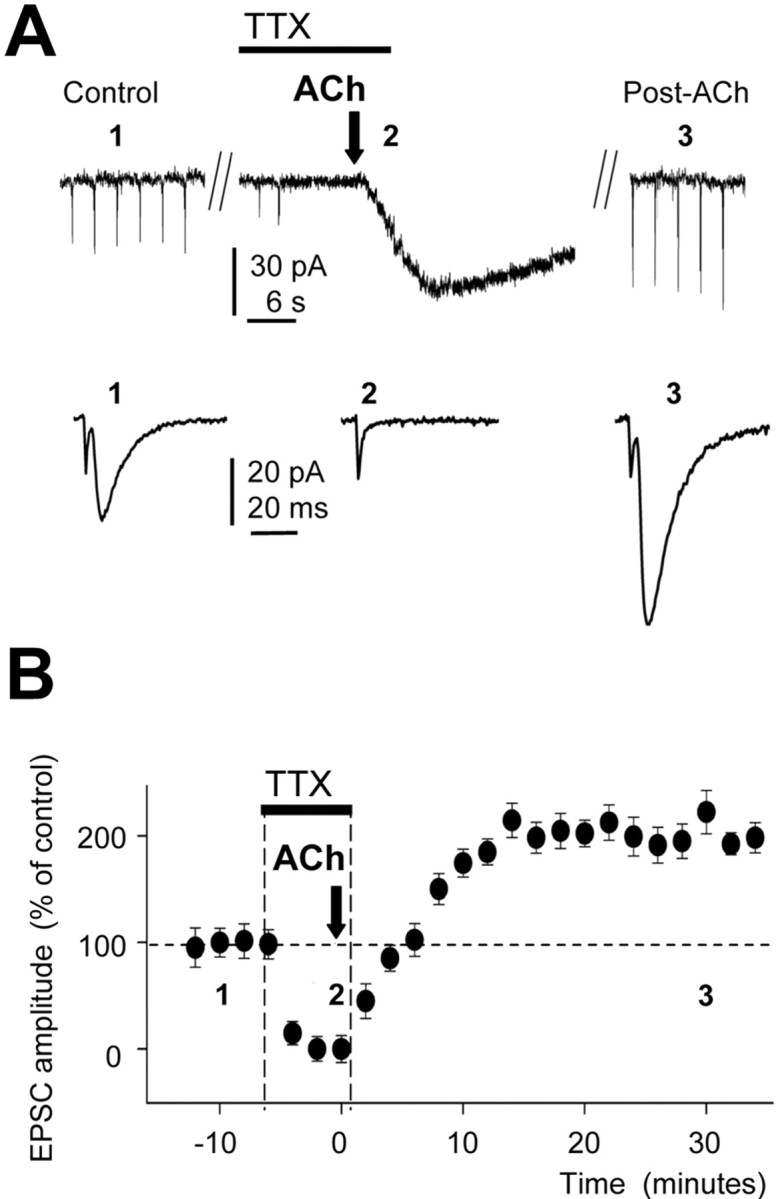
Cholinergic-glutamatergic interactions influence forms of synaptic plasticity that are thought to mediate memory and learning. We tested in vitro the induction of long-lasting synaptic enhancement at Schaffer collaterals by acetylcholine (ACh) at the apical dendrite of CA1 pyramidal neurons and in vivo by stimulation of cholinergic afferents. In vitro ACh induced a Ca2+ wave and synaptic enhancement mediated by insertion of AMPA receptors in spines. Activation of muscarinic ACh receptors (mAChRs) and Ca2+ release from inositol 1,4,5-trisphosphate (IP3)-sensitive stores were required for this synaptic enhancement that was insensitive to blockade of NMDA receptors and also triggered by IP3 uncaging. Activation of cholinergic afferents in vivo induced an analogous atropine-sensitive synaptic enhancement. We describe a novel form of synaptic enhancement (LTP(IP3)) that is induced in vitro and in vivo by activation of mAChRs. We conclude that Ca2+ released from postsynaptic endoplasmic reticulum stores is the critical event in the induction of this unique form of long-lasting synaptic enhancement.
Figures







References
-
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. - PubMed
-
- Aigner TG. Pharmacology of memory: cholinergic-glutamatergic interactions. Curr Opin Neurobiol. 1995;5:155–160. - PubMed
-
- Amenta F, Liu A, Giannella M, Pigini M, Tayebati SK, Zaccheo D. Age-related changes in the density of muscarinic cholinergic M1 and M2 receptor subtypes in pyramidal neurons of the rat hippocampus. Eur J Histochem. 1995;39:107–116. - PubMed
-
- Ashby MC, Ibaraki K, Henley JM. It's green outside: tracking cell surface proteins with pH-sensitive GFP. Trends Neurosci. 2004;27:257–261. - PubMed
-
- Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–2040. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous