

Neonatal diagnosis and treatment of Menkes disease
- PMID: 18256395
- PMCID: PMC3477514
- DOI: 10.1056/NEJMoa070613
Neonatal diagnosis and treatment of Menkes disease
Abstract
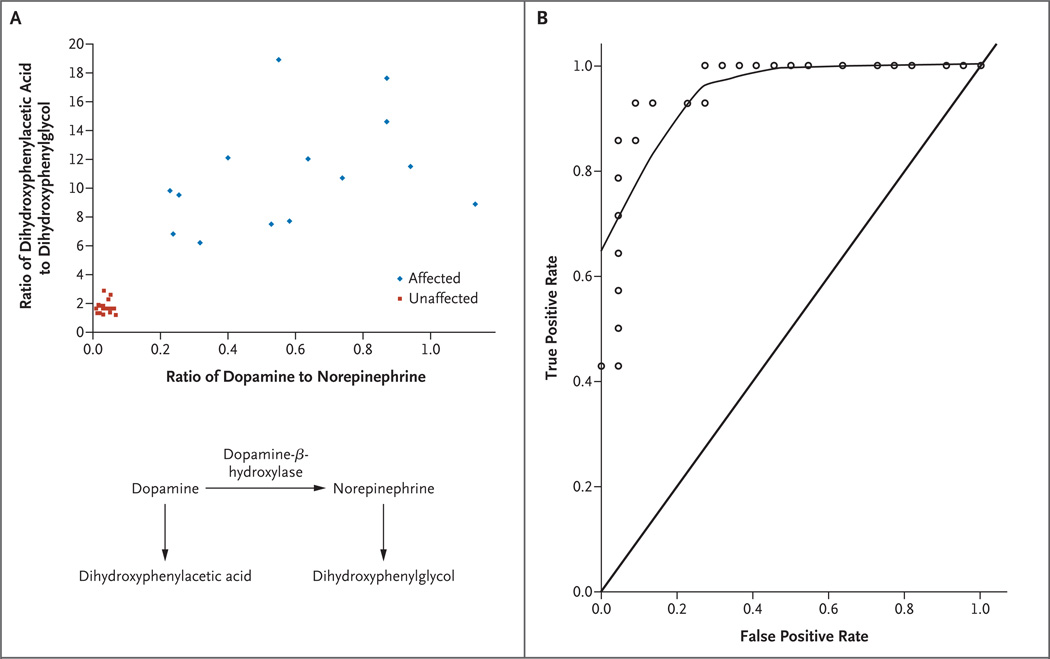
Background: Menkes disease is a fatal neurodegenerative disorder of infancy caused by diverse mutations in a copper-transport gene, ATP7A. Early treatment with copper injections may prevent death and illness, but presymptomatic detection is hindered by the inadequate sensitivity and specificity of diagnostic tests. Exploiting the deficiency of a copper enzyme, dopamine-beta-hydroxylase, we prospectively evaluated the diagnostic usefulness of plasma neurochemical levels, assessed the clinical effect of early detection, and investigated the molecular bases for treatment outcomes.
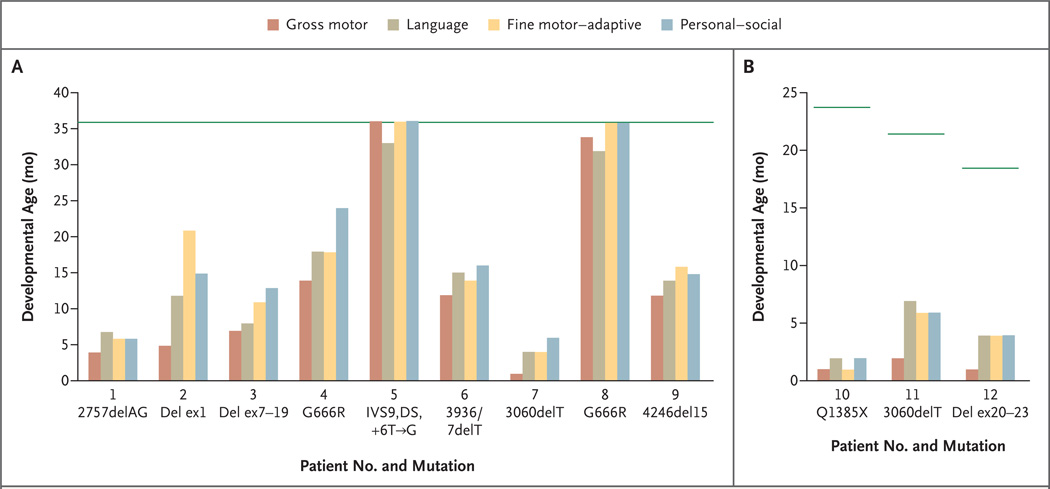
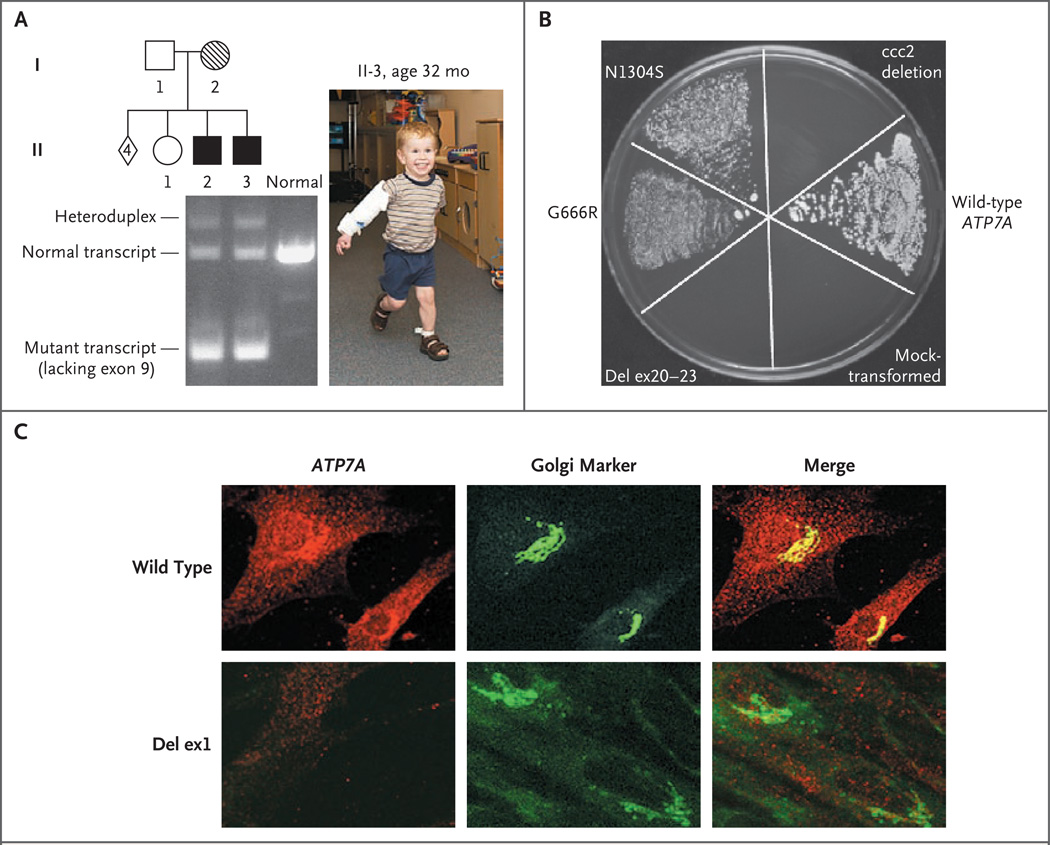
Methods: Between May 1997 and July 2005, we measured plasma dopamine, norepinephrine, dihydroxyphenylacetic acid, and dihydroxyphenylglycol in 81 infants at risk. In 12 newborns who met the eligibility criteria and began copper-replacement therapy within 22 days after birth, we tracked survival and neurodevelopment longitudinally for 1.5 to 8 years. We characterized ATP7A mutations using yeast complementation, reverse-transcriptase-polymerase-chain-reaction analysis, and immunohistochemical analysis.
Results: Of 81 infants at risk, 46 had abnormal neurochemical findings indicating low dopamine-beta-hydroxylase activity. On the basis of longitudinal follow-up, patients were classified as affected or unaffected by Menkes disease, and the neurochemical profiles were shown to have high sensitivity and specificity for detecting disease. Among 12 newborns with positive screening tests who were treated early with copper, survival at a median follow-up of 4.6 years was 92%, as compared with 13% at a median follow-up of 1.8 years for a historical control group of 15 late-diagnosis and late-treatment patients. Two of the 12 patients had normal neurodevelopment and brain myelination; 1 of these patients had a mutation that complemented a Saccharomyces cerevisiae copper-transport mutation, indicating partial ATPase activity, and the other had a mutation that allowed some correct ATP7A splicing.
Conclusions: Neonatal diagnosis of Menkes disease by plasma neurochemical measurements and early treatment with copper may improve clinical outcomes. Affected newborns who have mutations that do not completely abrogate ATP7A function may be especially responsive to early copper treatment. (ClinicalTrials.gov number, NCT00001262.)
Copyright 2008 Massachusetts Medical Society.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
References
-
- Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7–13. [Erratum, Nat Genet 1993;3:273.] - PubMed
-
- Chelly J, Tümer Z, Tønnesen T, et al. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat Genet. 1993;3:14–19. - PubMed
-
- Mercer JFB, Livingston J, Hall B, et al. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat Genet. 1993;3:20–25. - PubMed
-
- Kaler SG. Menkes disease. Adv Pediatr. 1994;41:263–304. [Erratum, Adv Pediatr 1995;42:xxxi.] - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous