

Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets
- PMID: 18258921
- PMCID: PMC2259103
- DOI: 10.1101/gr.7080508
Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets
Abstract
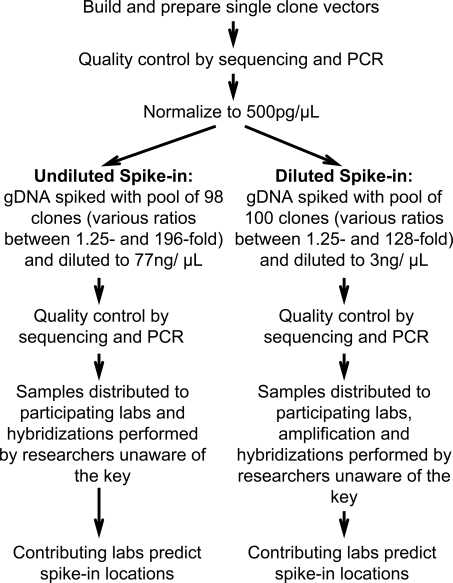
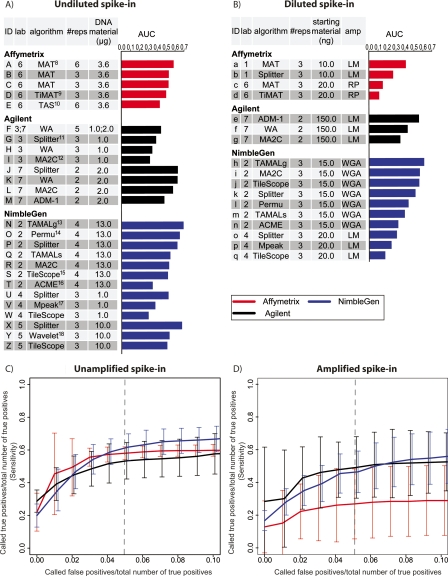
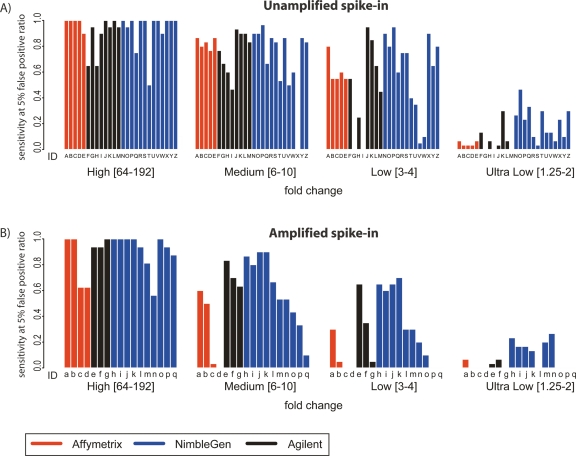
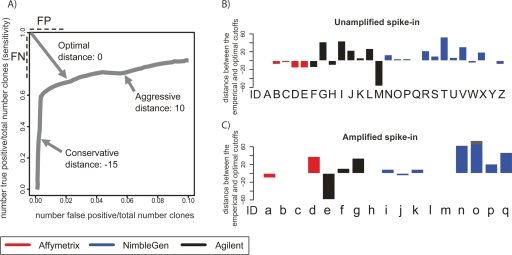
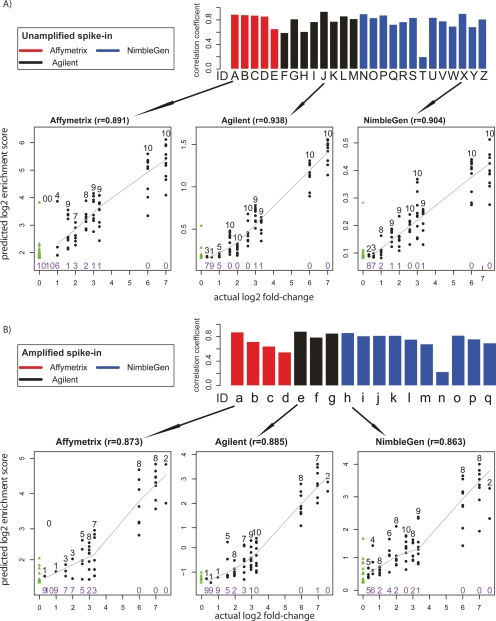
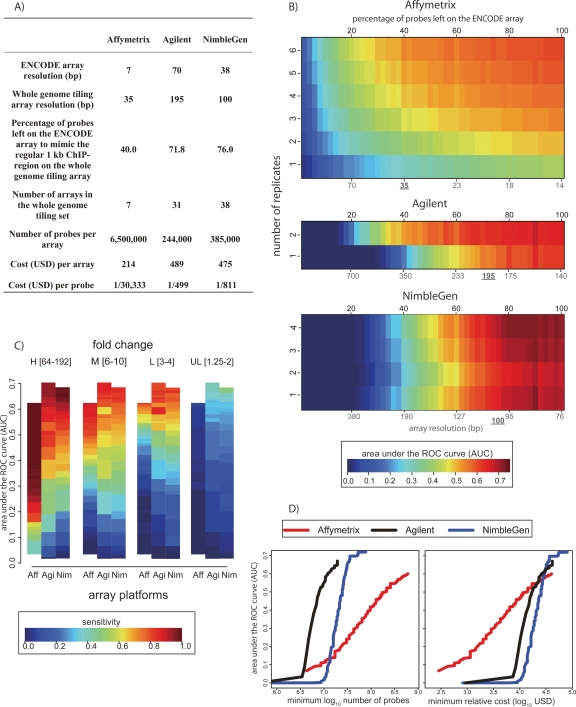
The most widely used method for detecting genome-wide protein-DNA interactions is chromatin immunoprecipitation on tiling microarrays, commonly known as ChIP-chip. Here, we conducted the first objective analysis of tiling array platforms, amplification procedures, and signal detection algorithms in a simulated ChIP-chip experiment. Mixtures of human genomic DNA and "spike-ins" comprised of nearly 100 human sequences at various concentrations were hybridized to four tiling array platforms by eight independent groups. Blind to the number of spike-ins, their locations, and the range of concentrations, each group made predictions of the spike-in locations. We found that microarray platform choice is not the primary determinant of overall performance. In fact, variation in performance between labs, protocols, and algorithms within the same array platform was greater than the variation in performance between array platforms. However, each array platform had unique performance characteristics that varied with tiling resolution and the number of replicates, which have implications for cost versus detection power. Long oligonucleotide arrays were slightly more sensitive at detecting very low enrichment. On all platforms, simple sequence repeats and genome redundancy tended to result in false positives. LM-PCR and WGA, the most popular sample amplification techniques, reproduced relative enrichment levels with high fidelity. Performance among signal detection algorithms was heavily dependent on array platform. The spike-in DNA samples and the data presented here provide a stable benchmark against which future ChIP platforms, protocol improvements, and analysis methods can be evaluated.
Figures
References
-
- Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Massa H.F., Trask B.J., Eichler E.E., Trask B.J., Eichler E.E., Eichler E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. - PMC - PubMed
-
- Bieda M., Xu X., Singer M.A., Green R., Farnham P.J., Xu X., Singer M.A., Green R., Farnham P.J., Singer M.A., Green R., Farnham P.J., Green R., Farnham P.J., Farnham P.J. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. - PMC - PubMed
-
- Carroll J.S., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Keeton E.K., Fertuck K.C., Hall G.F., Fertuck K.C., Hall G.F., Hall G.F., et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. - PubMed
-
- Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Sementchenko V., Cheng J., Williams A.J., Cheng J., Williams A.J., Williams A.J., et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases