

High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression
- PMID: 18263775
- PMCID: PMC2276441
- DOI: 10.1105/tpc.107.056879
High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression
Abstract
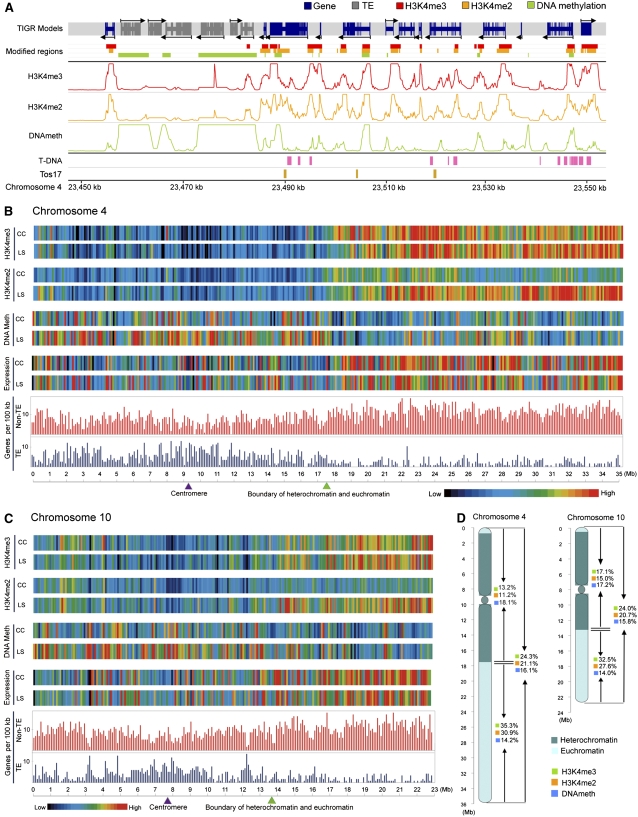
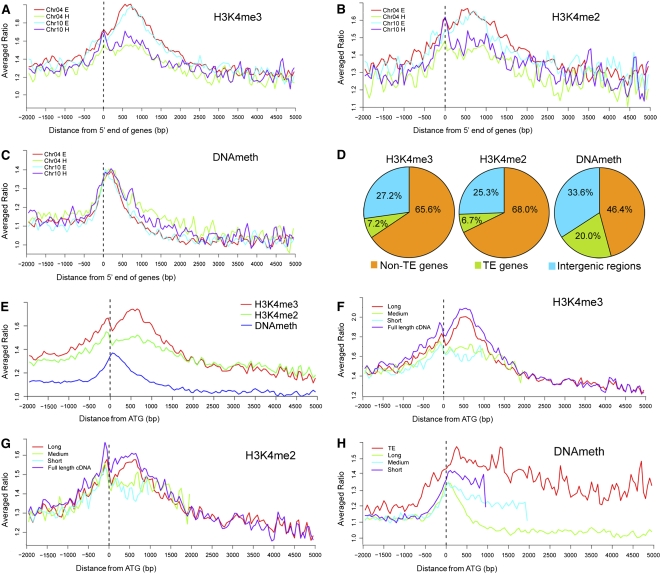
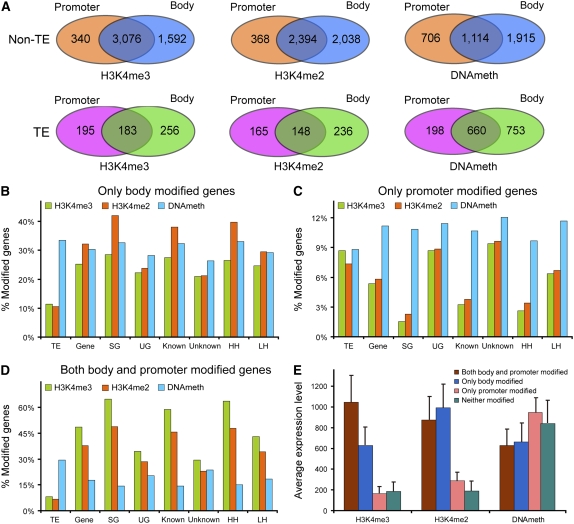
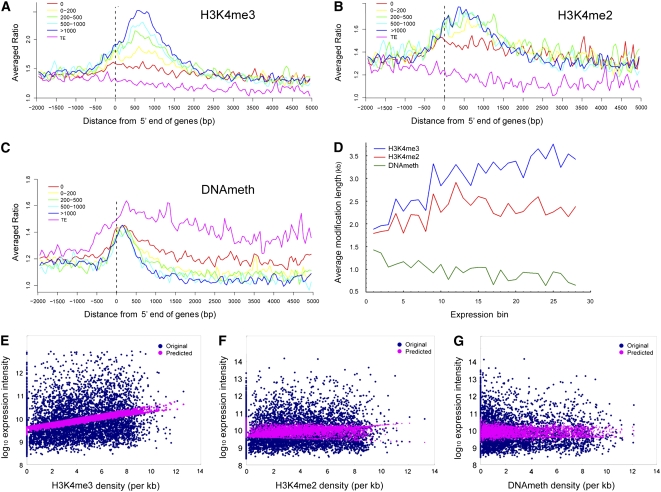
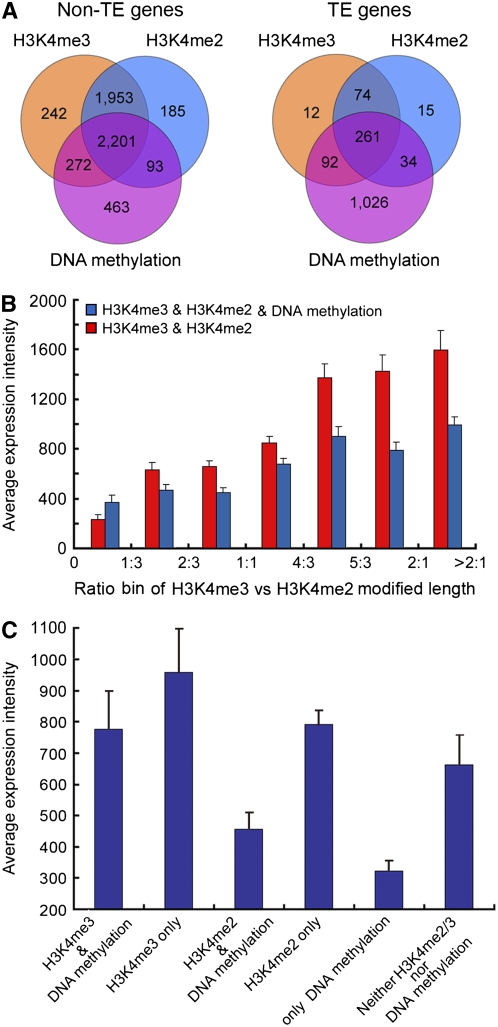
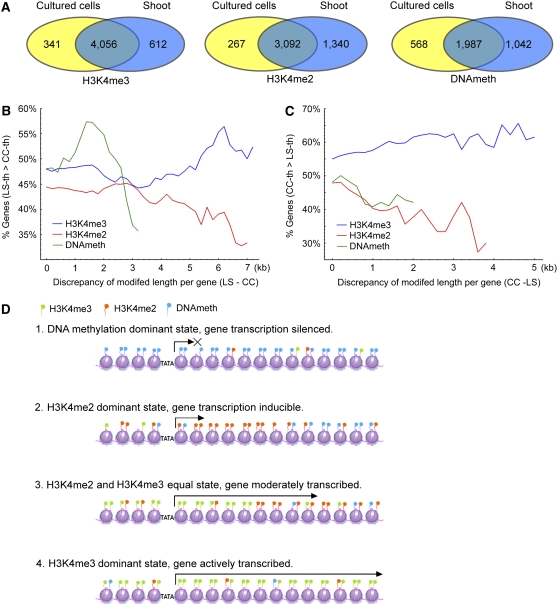
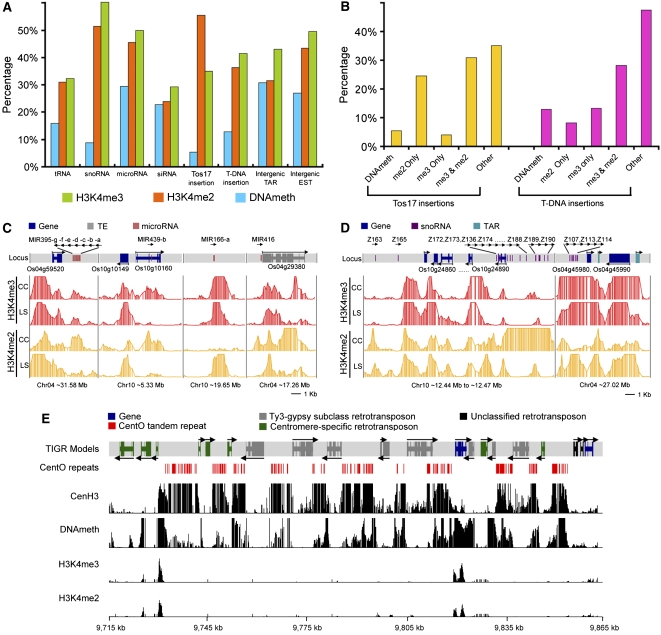
We present high-resolution maps of DNA methylation and H3K4 di- and trimethylation of two entire chromosomes and two fully sequenced centromeres in rice (Oryza sativa) shoots and cultured cells. This analysis reveals combinatorial interactions between these epigenetic modifications and chromatin structure and gene expression. Cytologically densely stained heterochromatin had less H3K4me2 and H3K4me3 and more methylated DNA than the less densely stained euchromatin, whereas centromeres had a unique epigenetic composition. Most transposable elements had highly methylated DNA but no H3K4 methylation, whereas more than half of protein-coding genes had both methylated DNA and di- and/or trimethylated H3K4. Methylation of DNA but not H3K4 was correlated with suppressed transcription. By contrast, when both DNA and H3K4 were methylated, transcription was only slightly reduced. Transcriptional activity was positively correlated with the ratio of H3K4me3/H3K4me2: genes with predominantly H3K4me3 were actively transcribed, whereas genes with predominantly H3K4me2 were transcribed at moderate levels. More protein-coding genes contained all three modifications, and more transposons contained DNA methylation in shoots than cultured cells. Differential epigenetic modifications correlated to tissue-specific expression between shoots and cultured cells. Collectively, this study provides insights into the rice epigenomes and their effect on gene expression and plant development.
Figures
Similar articles
-
Integration of cytological features with molecular and epigenetic properties of rice chromosome 4.Mol Plant. 2008 Sep;1(5):816-29. doi: 10.1093/mp/ssn037. Epub 2008 Aug 8. Mol Plant. 2008. PMID: 19825584
-
Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana.Genome Biol. 2009;10(6):R62. doi: 10.1186/gb-2009-10-6-r62. Epub 2009 Jun 9. Genome Biol. 2009. PMID: 19508735 Free PMC article.
-
Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica.Mol Plant. 2013 Sep;6(5):1463-72. doi: 10.1093/mp/sst018. Epub 2013 Jan 25. Mol Plant. 2013. PMID: 23355544 Free PMC article.
-
Rice as a model for centromere and heterochromatin research.Chromosome Res. 2007;15(1):77-84. doi: 10.1007/s10577-006-1104-z. Chromosome Res. 2007. PMID: 17295128 Review.
-
Histone modification and the control of heterochromatic gene silencing in Drosophila.Chromosome Res. 2006;14(4):377-92. doi: 10.1007/s10577-006-1066-1. Chromosome Res. 2006. PMID: 16821134 Review.
Cited by
-
Cross-species analysis of genic GC3 content and DNA methylation patterns.Genome Biol Evol. 2013;5(8):1443-56. doi: 10.1093/gbe/evt103. Genome Biol Evol. 2013. PMID: 23833164 Free PMC article.
-
Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice.Plant Cell. 2012 Nov;24(11):4407-21. doi: 10.1105/tpc.112.102269. Epub 2012 Nov 13. Plant Cell. 2012. PMID: 23150632 Free PMC article.
-
EpiHRMAssay, in tube and in silico combined approach for the scanning and epityping of heterogeneous DNA methylation.Biol Methods Protoc. 2017 Jan 25;2(1):bpw008. doi: 10.1093/biomethods/bpw008. eCollection 2017 Jan. Biol Methods Protoc. 2017. PMID: 32161783 Free PMC article.
-
Epigenetic modifications in plants: an evolutionary perspective.Curr Opin Plant Biol. 2011 Apr;14(2):179-86. doi: 10.1016/j.pbi.2010.12.002. Epub 2011 Jan 11. Curr Opin Plant Biol. 2011. PMID: 21233005 Free PMC article. Review.
-
Leveraging histone modifications to improve genome annotations.G3 (Bethesda). 2021 Sep 27;11(10):jkab263. doi: 10.1093/g3journal/jkab263. G3 (Bethesda). 2021. PMID: 34568920 Free PMC article.
References
-
- Barski, A., Cuddapah, S., Cui, K., Roh, T.-Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129 823–837. - PubMed
-
- Bender, J. (2004). DNA methylation and epigenetics. Annu. Rev. Plant Biol. 55 41–68. - PubMed
-
- Bernstein, B.E., Kamal, M., Lindblad-Toh, K., Bekiranov, S., Bailey, D.K., Huebert, D.J., McMahon, S., Karlsson, E.K., Kulbokas III, E.J., Gingeras, T.R., Schreiber, S.L., and Lander, E.S. (2005). Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120 169–181. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
