

Mutations in FN1 cause glomerulopathy with fibronectin deposits
- PMID: 18268355
- PMCID: PMC2268172
- DOI: 10.1073/pnas.0707730105
Mutations in FN1 cause glomerulopathy with fibronectin deposits
Abstract
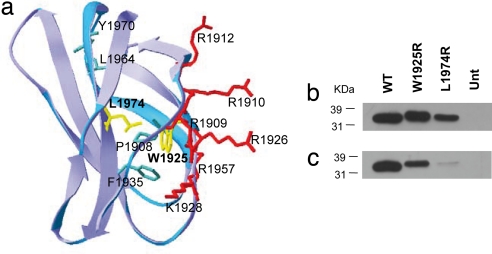
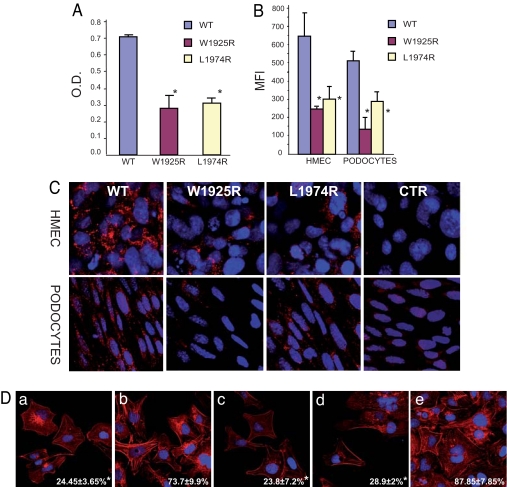
Glomerulopathy with fibronectin (FN) deposits (GFND) is an autosomal dominant disease with age-related penetrance, characterized by proteinuria, microscopic hematuria, hypertension, and massive glomerular deposits of FN that lead to end-stage renal failure. The genetic abnormality underlying GFND was still unknown. We hypothesized that mutations in FN1, which encodes FN, were the cause of GFND. In a large Italian pedigree with eight affected subjects, we found linkage with GFND at the FN1 locus at 2q32. We sequenced the FN1 in 15 unrelated pedigrees and found three heterozygous missense mutations, the W1925R, L1974R, and Y973C, that cosegregated with the disease in six pedigrees. The mutations affected two domains of FN (Hep-II domain for the W1925R and the L1974R, and Hep-III domain for the Y973C) that play key roles in FN-cell interaction and in FN fibrillogenesis. Mutant recombinant Hep-II fragments were expressed, and functional studies revealed a lower binding to heparin and to endothelial cells and podocytes compared with wild-type Hep-II and an impaired capability to induce endothelial cell spreading and cytoskeletal reorganization. Overall dominant mutations in FN1 accounted for 40% of cases of GFND in our study group. These findings may help understanding the pathogenesis of proteinuria and glomerular FN deposits in GFND and possibly in more common renal diseases such as diabetic nephropathy, IgA nephropathy, and lupus nephritis. To our knowledge no FN1 mutation causing a human disease was previously reported.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Strom EH, Banfi G, Krapf R, Abt AB, Mazzucco G, Monga G, Gloor F, Neuweiler J, Riess R, Stosiek P, et al. Kidney Int. 1995;48:163–170. - PubMed
-
- Kornblihtt AR, Pesce CG, Alonso CR, Cramer P, Srebrow A, Werbajh S, Muro AF. FASEB J. 1996;10:248–257. - PubMed
-
- Hildebrandt F, Strahm B, Prochoroff A, Cybulla M, Gemperle O, Krapf R, Brandis M. Am J Med Genet. 1996;63:323–327. - PubMed
-
- Zhang Z, Kundu GC, Yuan CJ, Ward JM, Lee EJ, DeMayo F, Westphal H, Mukherjee AB. Science. 1997;276:1408–1412. - PubMed
-
- Vollmer M, Krapf R, Hildebrandt F. Nephrol Dial Transplant. 1998;13:2417–2418. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
