

Multiple osteochondromas
- PMID: 18271966
- PMCID: PMC2276198
- DOI: 10.1186/1750-1172-3-3
Multiple osteochondromas
Abstract
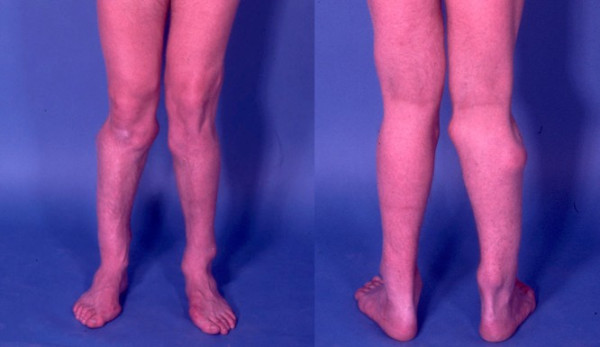
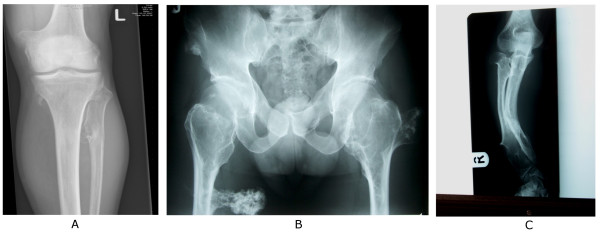
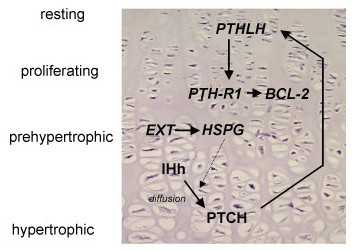
Multiple osteochondromas (MO) is characterised by development of two or more cartilage capped bony outgrowths (osteochondromas) of the long bones. The prevalence is estimated at 1:50,000, and it seems to be higher in males (male-to-female ratio 1.5:1). Osteochondromas develop and increase in size in the first decade of life, ceasing to grow when the growth plates close at puberty. They are pedunculated or sessile (broad base) and can vary widely in size. The number of osteochondromas may vary significantly within and between families, the mean number of locations is 15-18. The majority are asymptomatic and located in bones that develop from cartilage, especially the long bones of the extremities, predominantly around the knee. The facial bones are not affected. Osteochondromas may cause pain, functional problems and deformities, especially of the forearm, that may be reason for surgical removal. The most important complication is malignant transformation of osteochondroma towards secondary peripheral chondrosarcoma, which is estimated to occur in 0.5-5%. MO is an autosomal dominant disorder and is genetically heterogeneous. In almost 90% of MO patients germline mutations in the tumour suppressor genes EXT1 or EXT2 are found. The EXT genes encode glycosyltransferases, catalyzing heparan sulphate polymerization. The diagnosis is based on radiological and clinical documentation, supplemented with, if available, histological evaluation of osteochondromas. If the exact mutation is known antenatal diagnosis is technically possible. MO should be distinguished from metachondromatosis, dysplasia epiphysealis hemimelica and Ollier disease. Osteochondromas are benign lesions and do not affect life expectancy. Management includes removal of osteochondromas when they give complaints. Removed osteochondromas should be examined for malignant transformation towards secondary peripheral chondrosarcoma. Patients should be well instructed and regular follow-up for early detection of malignancy seems justified. For secondary peripheral chondrosarcoma, en-bloc resection of the lesion and its pseudocapsule with tumour-free margins, preferably in a bone tumour referral centre, should be performed.
Figures
References
-
- Khurana J, Abdul-Karim F, Bovée JVMG. Osteochondroma. In: Fletcher CDM, Unni KK and Mertens F, editor. World Health Organization classification of tumours Pathology and genetics of tumours of soft tissue and bone. Lyon, IARC Press; 2002. pp. 234–236.
-
- Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer M. Incomplete penetrance and expressivity skewing in hereditary multiple exostoses. Clin Genet. 1997;52:12–16. - PubMed
-
- Bovée JVMG, Hogendoorn PCW. Multiple osteochondromas. In: Fletcher CDM, Unni KK and Mertens F, editor. World Health Organization classification of tumours Pathology and genetics of tumours of soft tissue and bone. Lyon, IARC Press; 2002. pp. 360–362.
-
- Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg [Am] 1994;76:986–992. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
