

Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways
- PMID: 18276609
- PMCID: PMC2902290
- DOI: 10.1093/hmg/ddn042
Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways
Abstract
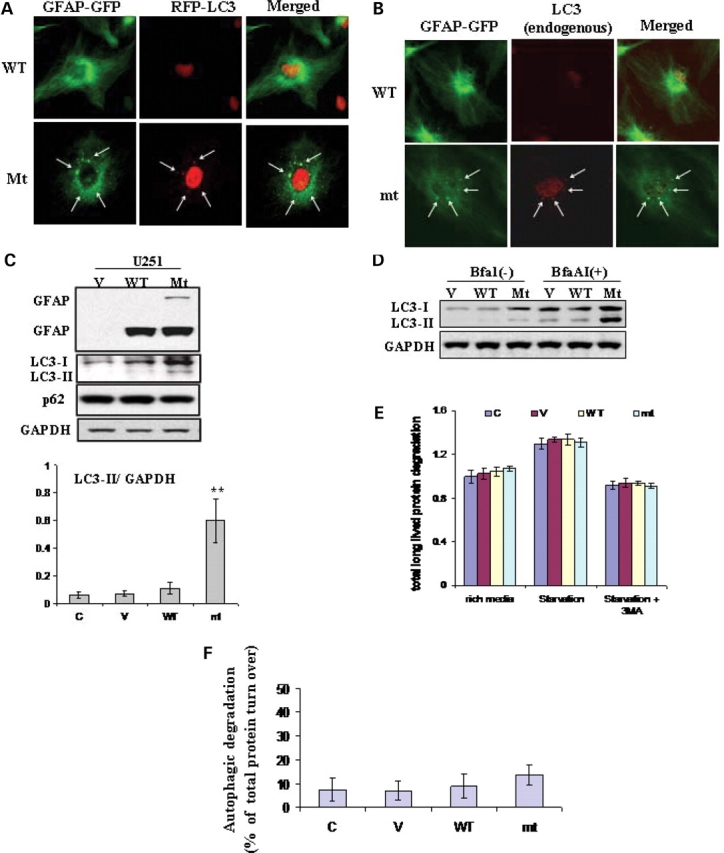
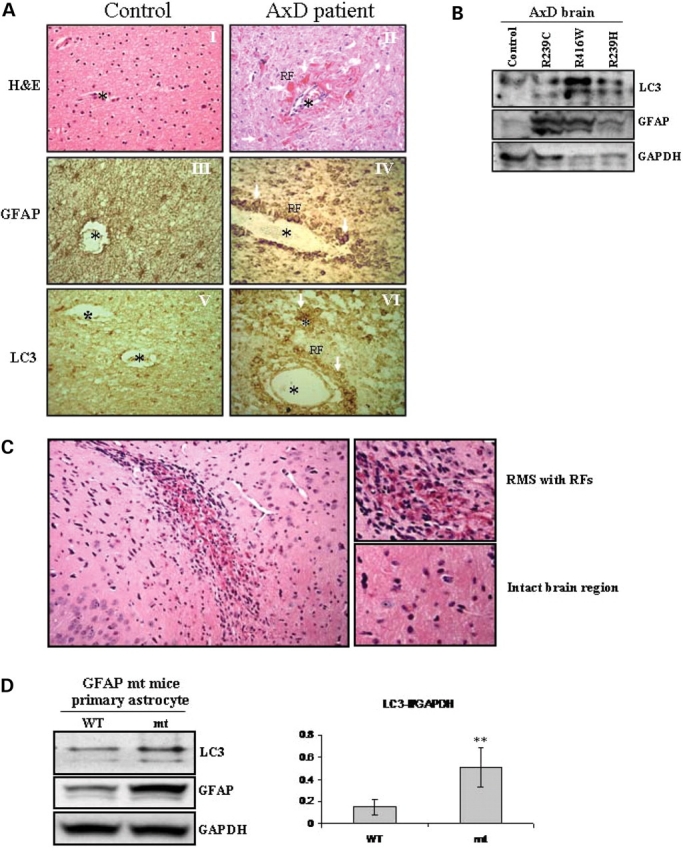
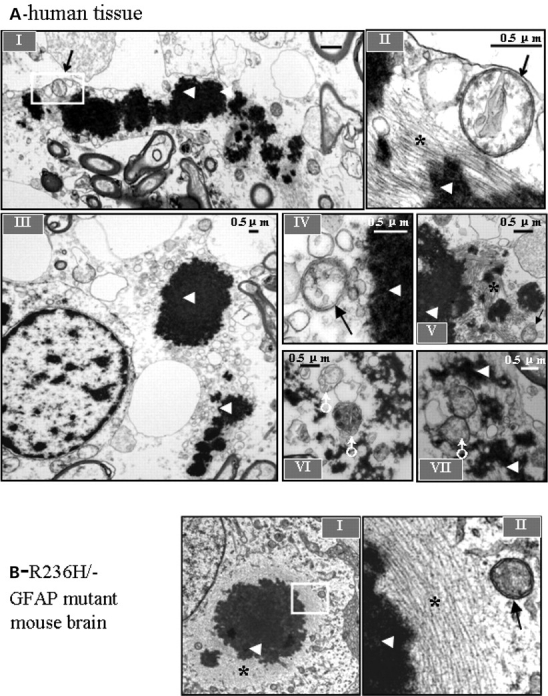
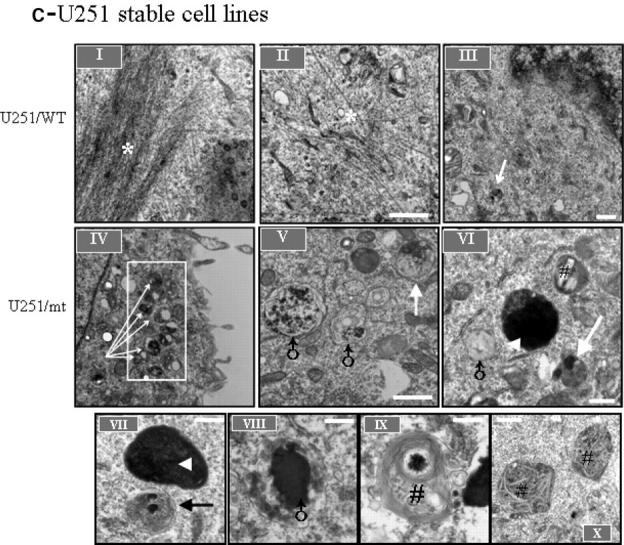
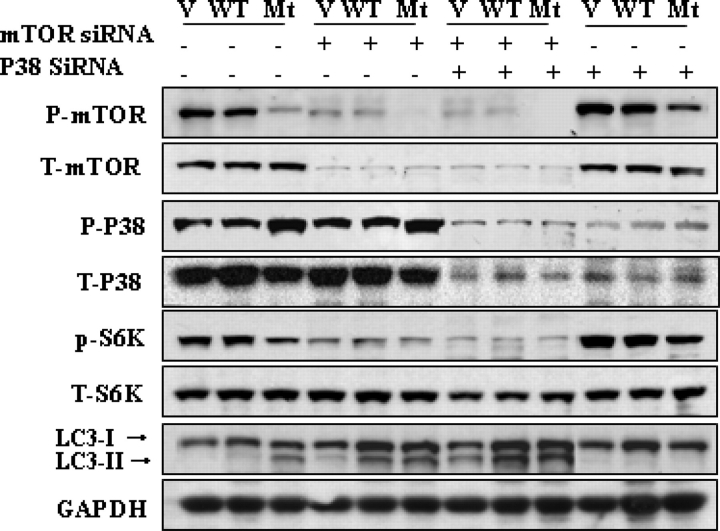
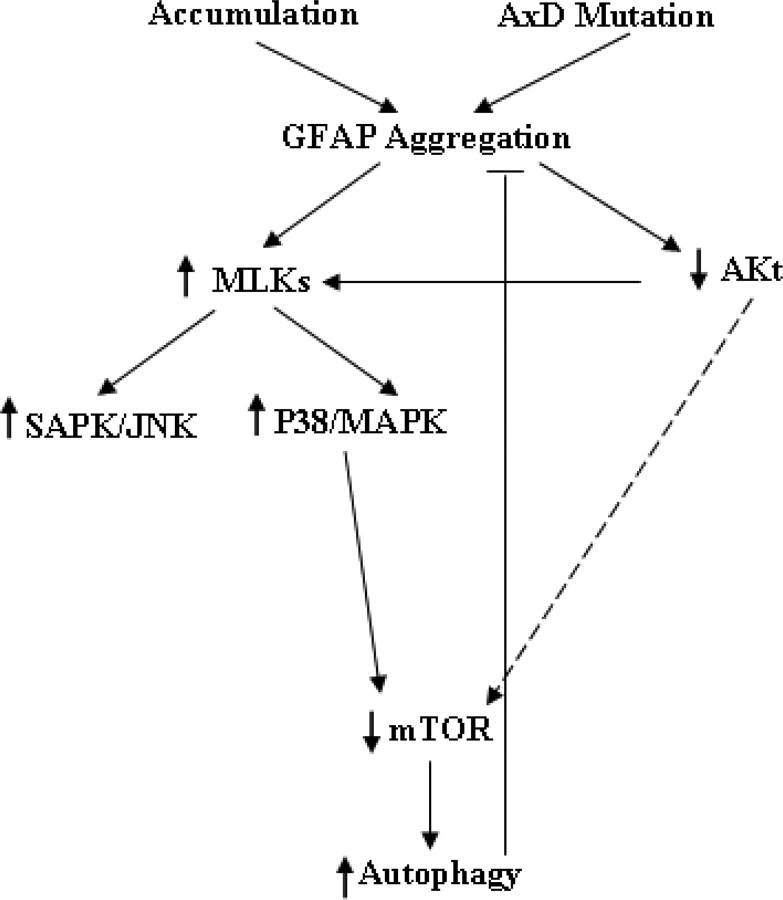
Glial fibrillary acidic protein (GFAP) is the principle intermediate filament (IF) protein in astrocytes. Mutations in the GFAP gene lead to Alexander disease (AxD), a rare, fatal neurological disorder characterized by the presence of abnormal astrocytes that contain GFAP protein aggregates, termed Rosenthal fibers (RFs), and the loss of myelin. All GFAP mutations cause the same histopathological defect, i.e. RFs, though little is known how the mutations affect protein accumulation as well as astrocyte function. In this study, we found that GFAP accumulation induces macroautophagy, a key clearance mechanism for prevention of aggregated proteins. This autophagic response is negatively regulated by mammalian target of rapamycin (mTOR). The activation of p38 MAPK by GFAP accumulation is in part responsible for the down-regulation of phosphorylated-mTOR and the subsequent activation of autophagy. Our study suggests that AxD mutant GFAP accumulation stimulates autophagy, in a manner regulated by p38 MAPK and mTOR signaling pathways. Autophagy, in turn, serves as a mechanism to reduce GFAP levels.
Figures
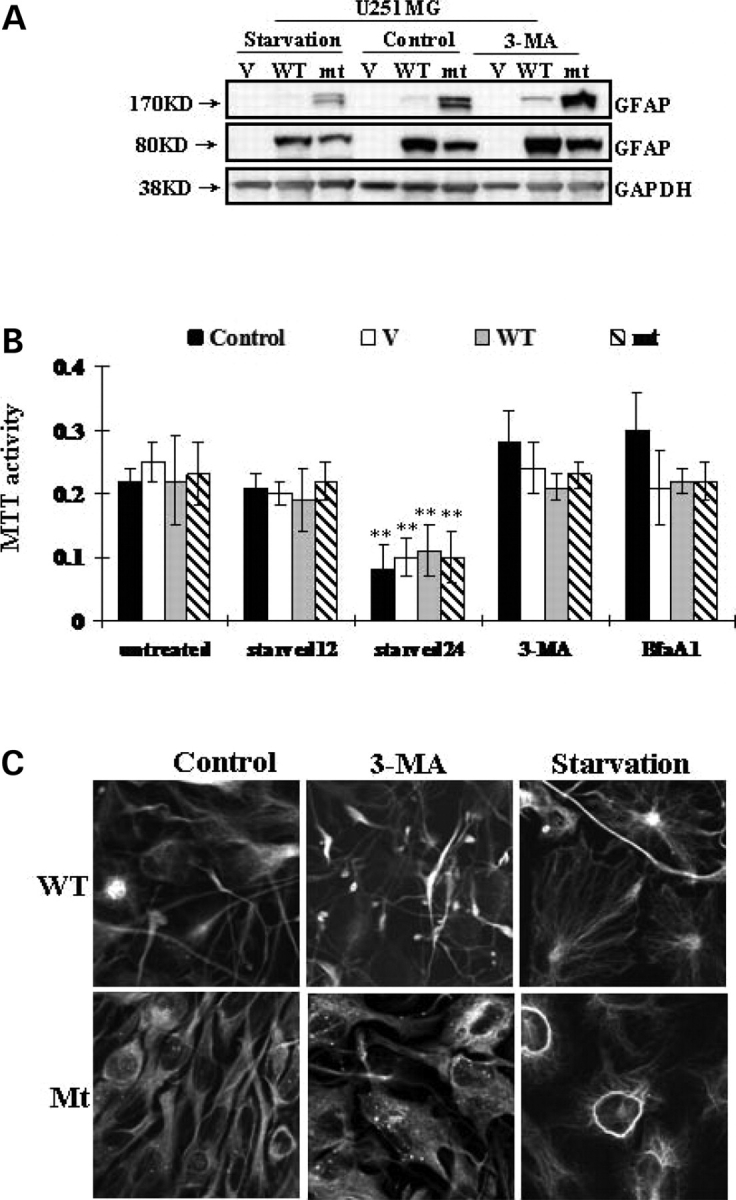
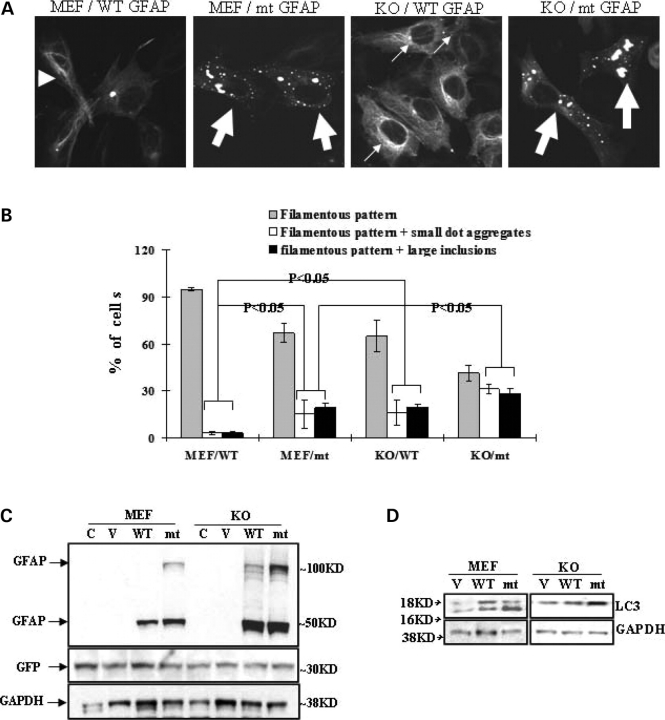
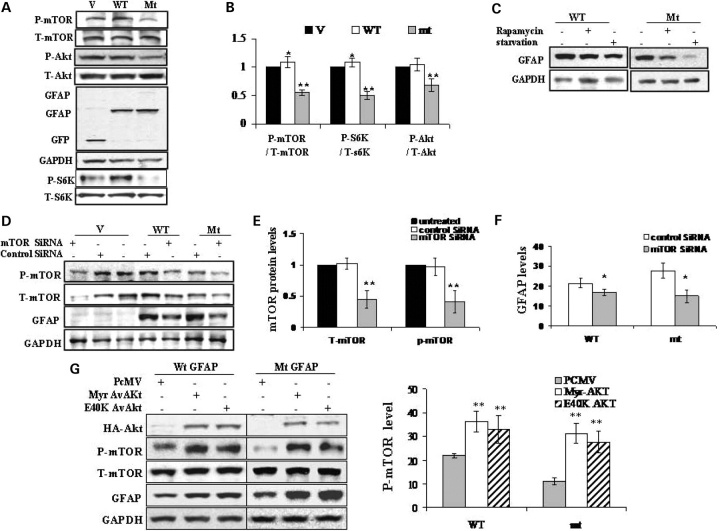
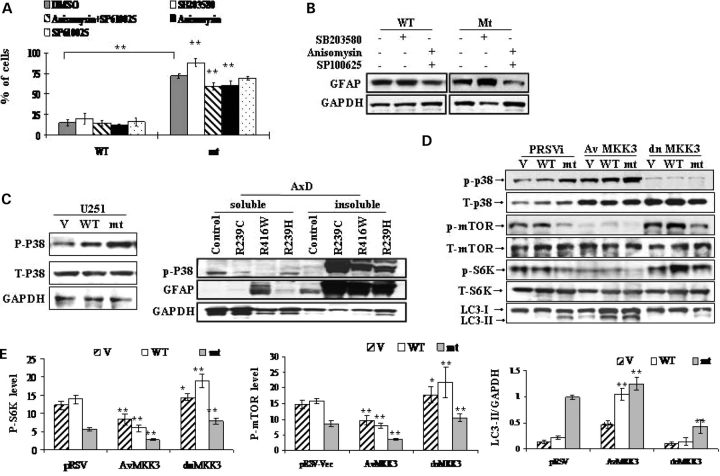
References
-
- Brenner M., Johnson A.B., Boespflug-Tanguy O., Rodriguez D., Goldman J.E., Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 2001;27:117–120. - PubMed
-
- Johnson A.B. Alexander disease: a leukodystrophy caused by a mutation in GFAP. Neurochem. Res. 2004;29:961–964. - PubMed
-
- Li R., Messing A., Goldman J.E., Brenner M. GFAP mutations in Alexander disease. Int. J. Dev. Neurosci. 2002;20:259–268. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
