

Accurate prediction of protein-protein interactions from sequence alignments using a Bayesian method
- PMID: 18277381
- PMCID: PMC2267735
- DOI: 10.1038/msb4100203
Accurate prediction of protein-protein interactions from sequence alignments using a Bayesian method
Abstract
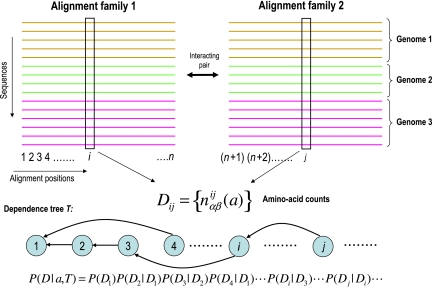
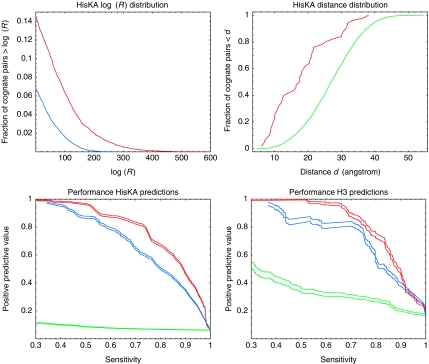
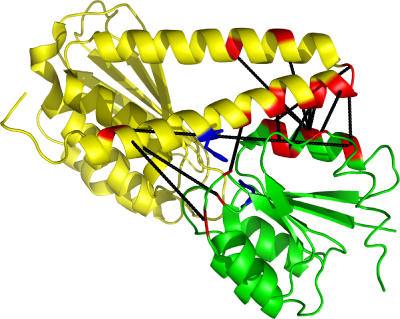
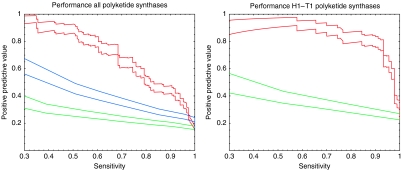
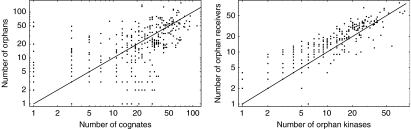
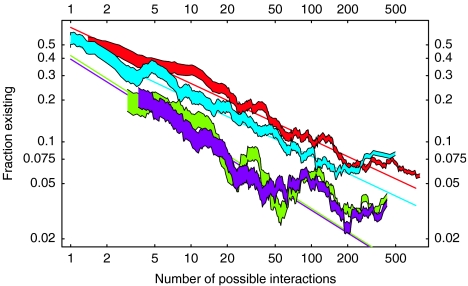
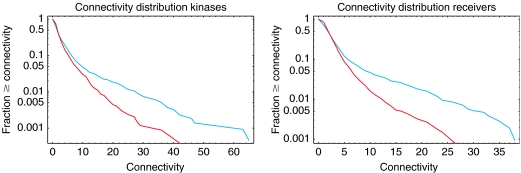
Accurate and large-scale prediction of protein-protein interactions directly from amino-acid sequences is one of the great challenges in computational biology. Here we present a new Bayesian network method that predicts interaction partners using only multiple alignments of amino-acid sequences of interacting protein domains, without tunable parameters, and without the need for any training examples. We first apply the method to bacterial two-component systems and comprehensively reconstruct two-component signaling networks across all sequenced bacteria. Comparisons of our predictions with known interactions show that our method infers interaction partners genome-wide with high accuracy. To demonstrate the general applicability of our method we show that it also accurately predicts interaction partners in a recent dataset of polyketide synthases. Analysis of the predicted genome-wide two-component signaling networks shows that cognates (interacting kinase/regulator pairs, which lie adjacent on the genome) and orphans (which lie isolated) form two relatively independent components of the signaling network in each genome. In addition, while most genes are predicted to have only a small number of interaction partners, we find that 10% of orphans form a separate class of 'hub' nodes that distribute and integrate signals to and from up to tens of different interaction partners.
Figures
References
-
- Ausmees N, Jacobs-Wagner C (2003) Spatial and temporal control of differentiation and cell cycle progression in Caulobacter crescentus. Annu Rev Microbiol 57: 225–247 - PubMed
-
- Biondi E, Reisinger S, Skerker J, Arif M, Perchuk B, Ryan K, Laub M (2006) Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature 444: 899–904 - PubMed
-
- Bork P, Jensen L, von Mering C, Ramani A, Lee I, Marcotte E (2004) Protein interaction networks from yeast to human. Curr Opin Struct Biol 14: 292–299 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
