

Application of CYP3A4 in vitro data to predict clinical drug-drug interactions; predictions of compounds as objects of interaction
- PMID: 18279465
- PMCID: PMC2432478
- DOI: 10.1111/j.1365-2125.2007.03070.x
Application of CYP3A4 in vitro data to predict clinical drug-drug interactions; predictions of compounds as objects of interaction
Abstract
What is already known about this subject: Numerous retrospective analyses have shown the utility of in vitro systems for predicting potential drug-drug interactions (DDIs). Prediction of DDIs from in vitro data is commonly obtained using estimates of enzyme K(i), inhibitor and substrate concentrations and absorption rate for substrate and inhibitor.
What this study adds: Using a generic approach for all test compounds, the findings from the current study showed the use of recombinant P450s provide a more robust in vitro measure of P450 contribution (fraction metabolized, f(m)) than that achieved when using chemical inhibitors in combination with human liver microsomes, for the prediction of potential CYP3A4 drug-drug interactions prior to clinical investigation. The current study supported the use of SIMCYP(R), a modelling and simulation software in utilizing the in vitro measures in the prediction of potential drug-drug interactions.
Aims: The aim of this study was to explore and optimize the in vitro and in silico approaches used for predicting clinical DDIs. A data set containing clinical information on the interaction of 20 Pfizer compounds with ketoconazole was used to assess the success of the techniques.
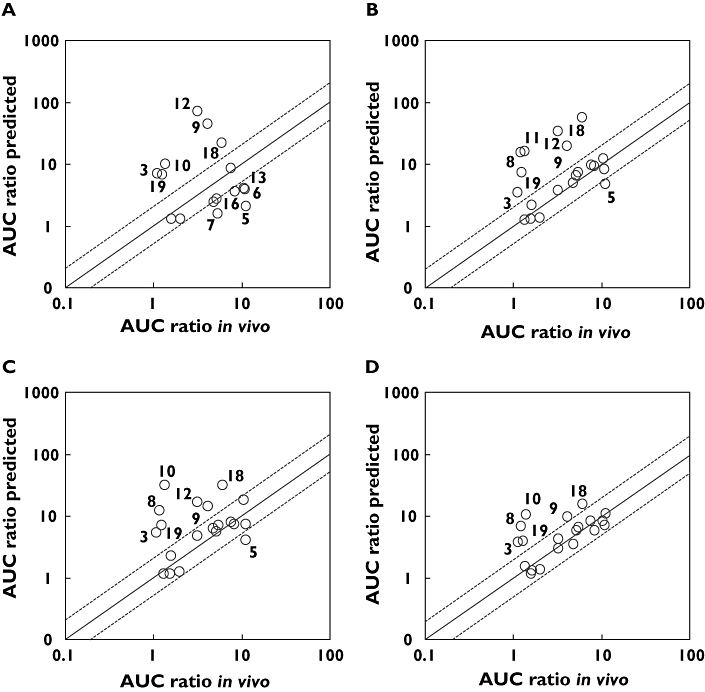
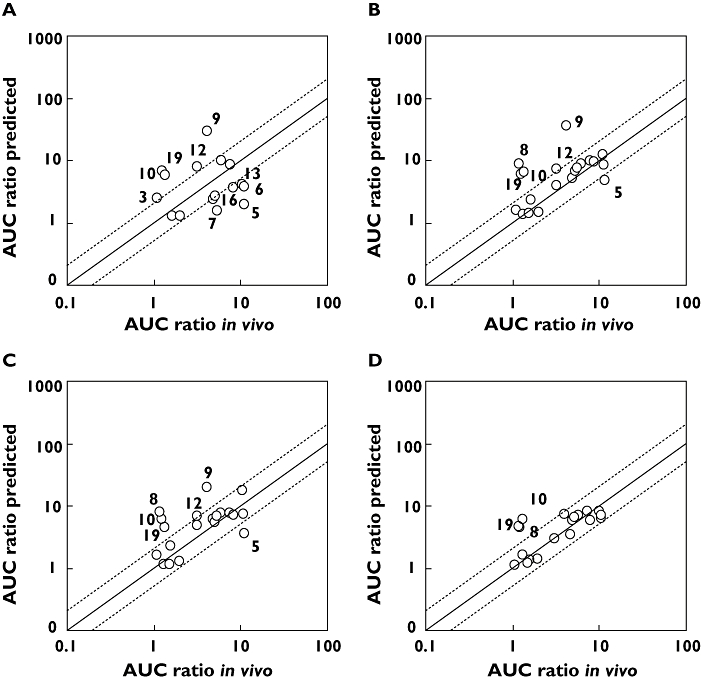
Methods: The study calculated the fraction and the rate of metabolism of 20 Pfizer compounds via each cytochrome P450. Two approaches were used to determine fraction metabolized (f(m)); 1) by measuring substrate loss in human liver microsomes (HLM) in the presence and absence of specific chemical inhibitors and 2) by measuring substrate loss in individual cDNA expressed P450s (also referred to as recombinant P450s (rhCYP)) The fractions metabolized via each CYP were used to predict the drug-drug interaction due to CYP3A4 inhibition by ketoconazole using the modelling and simulation software SIMCYP.
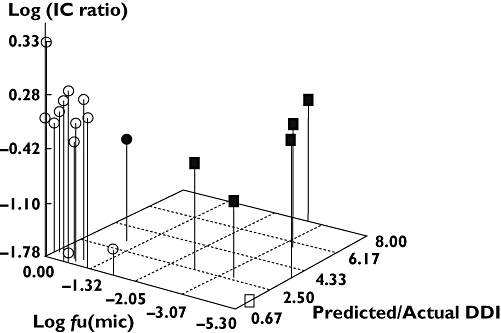
Results: When in vitro data were generated using Gentest supersomes, 85% of predictions were within two-fold of the observed clinical interaction. Using PanVera baculosomes, 70% of predictions were predicted within two-fold. In contrast using chemical inhibitors the accuracy was lower, predicting only 37% of compounds within two-fold of the clinical value. Poorly predicted compounds were found to either be metabolically stable and/or have high microsomal protein binding. The use of equilibrium dialysis to generate accurate protein binding measurements was especially important for highly bound drugs.
Conclusions: The current study demonstrated that the use of rhCYPs with SIMCYP provides a robust in vitro system for predicting the likelihood and magnitude of changes in clinical exposure of compounds as a consequence of CYP3A4 inhibition by a concomitantly administered drug.
Figures
References
-
- Shou M. Prediction of pharmacokinetics and drug–drug interactions from in vitro metabolism data. Curr Opin Drug Discov Devel. 2005;8:66–77. - PubMed
-
- Andersson TB, Bredberg E, Ericsson H, Sjoberg H. An evaluation of the in vitro metabolism data for predicting the clearance and drug–drug interaction potential of CYP2C9 substrates. Drug Metab Dispos. 2004;32:715–21. - PubMed
-
- McGinnity DF, Riley RJ. Predicting drug pharmacokinetics in humans from in vitro metabolism studies. Biochem Soc Trans. 2001;29:135–9. - PubMed
-
- Gibbs JP, Hyland R, Youdim K. Minimizing polymorphic metabolism in drug discovery: evaluation of the utility of in vitro methods for predicting pharmacokinetic consequences associated with CYP2D6 metabolism. Drug Metab Dispos. 2006;8:8. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
