

Rescoring docking hit lists for model cavity sites: predictions and experimental testing
- PMID: 18280498
- PMCID: PMC2752715
- DOI: 10.1016/j.jmb.2008.01.049
Rescoring docking hit lists for model cavity sites: predictions and experimental testing
Abstract
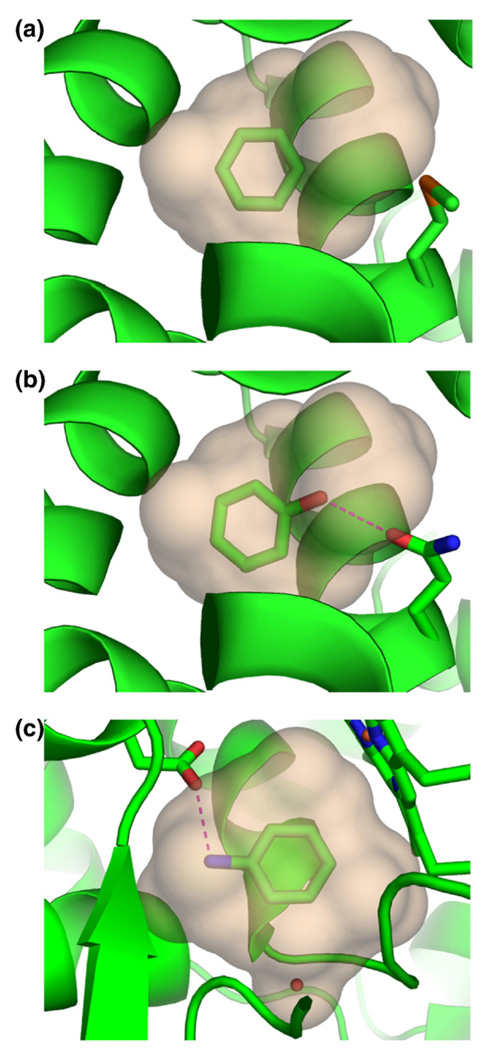
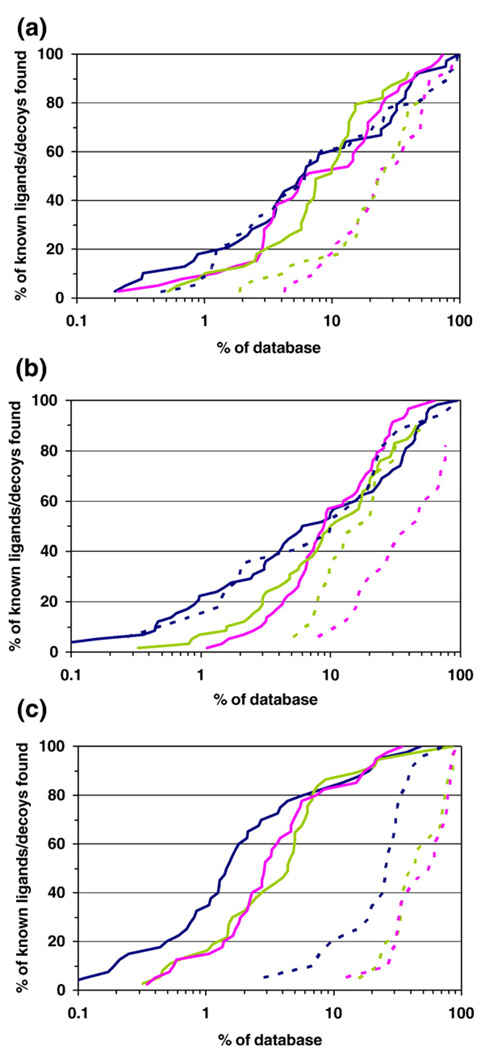
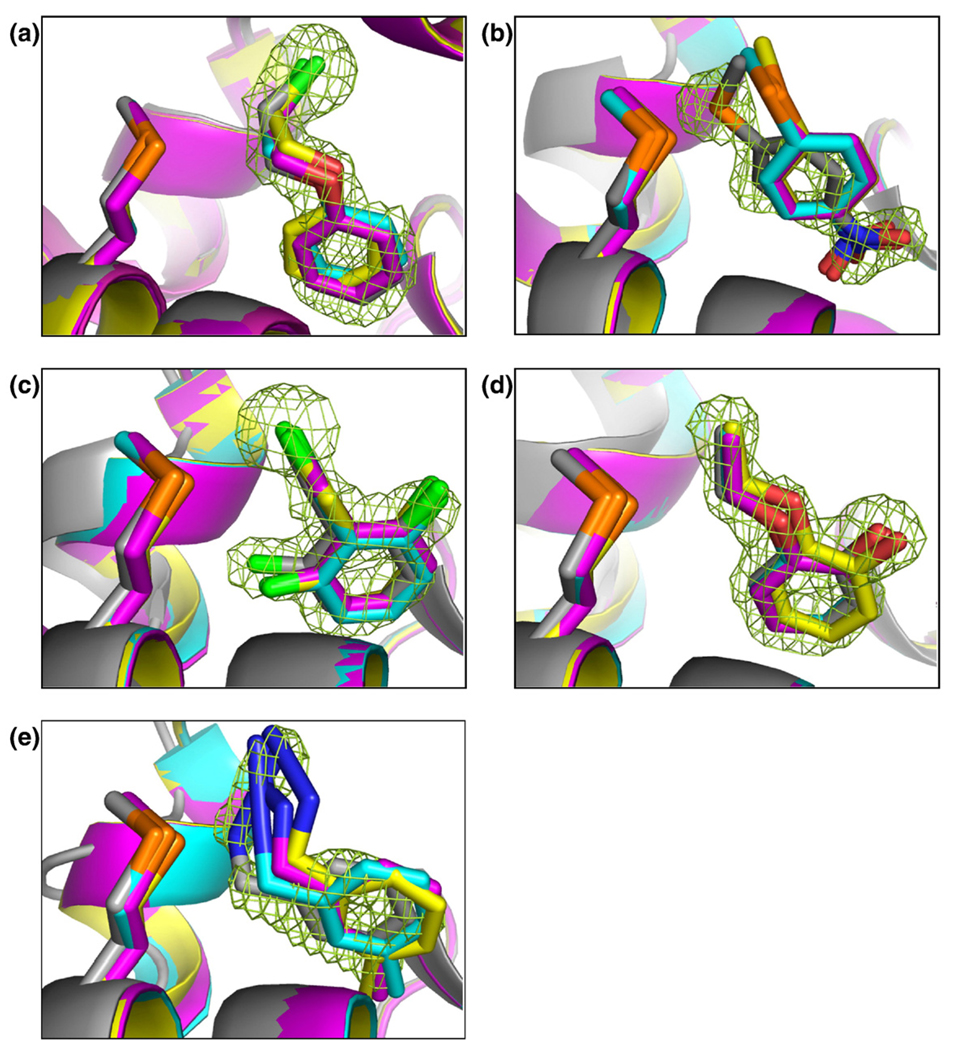
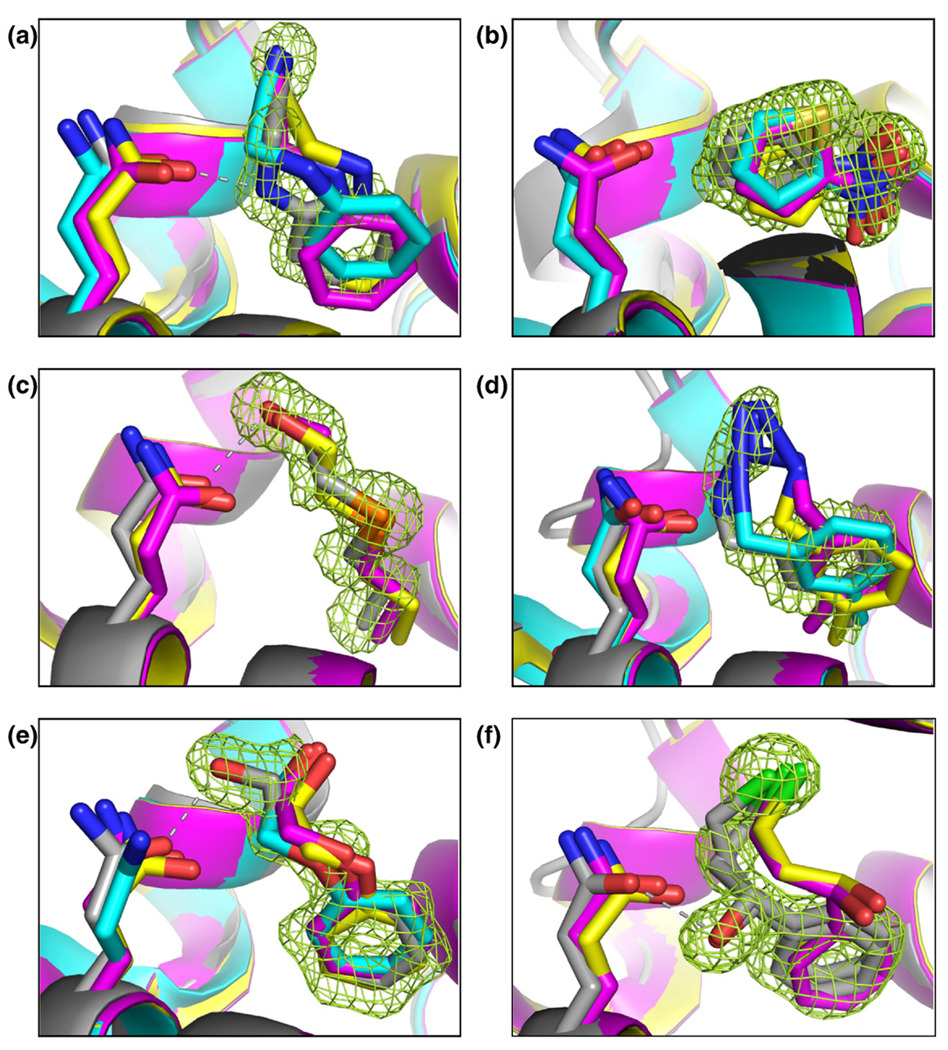
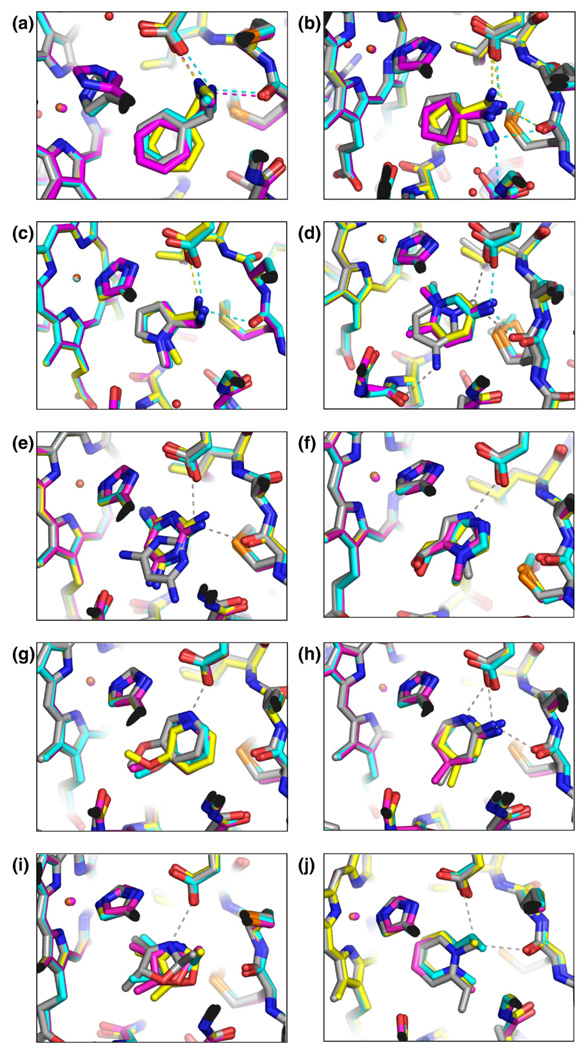
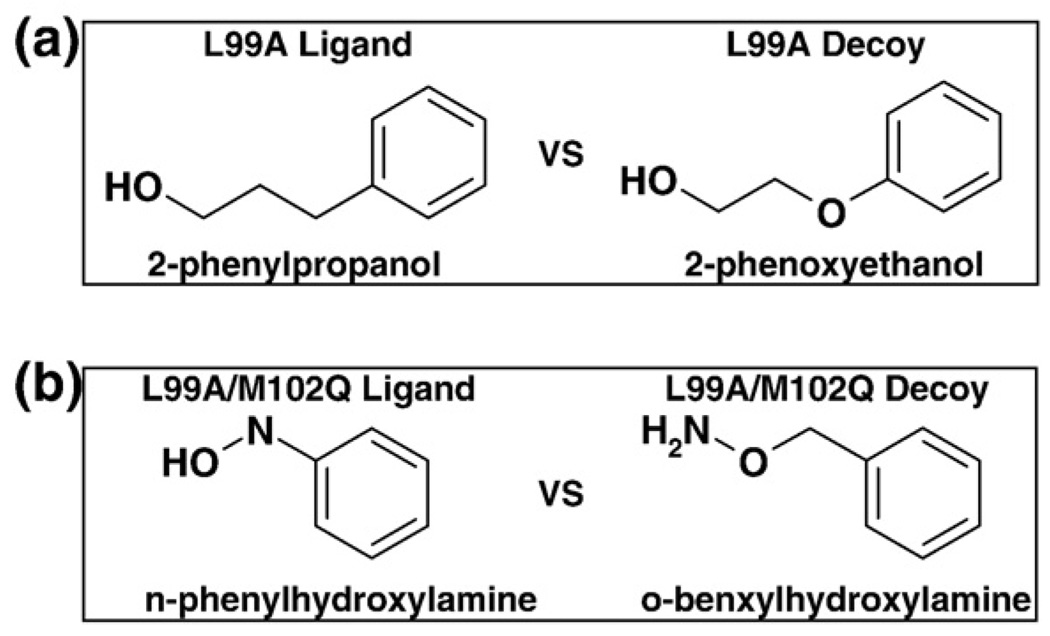
Molecular docking computationally screens thousands to millions of organic molecules against protein structures, looking for those with complementary fits. Many approximations are made, often resulting in low "hit rates." A strategy to overcome these approximations is to rescore top-ranked docked molecules using a better but slower method. One such is afforded by molecular mechanics-generalized Born surface area (MM-GBSA) techniques. These more physically realistic methods have improved models for solvation and electrostatic interactions and conformational change compared to most docking programs. To investigate MM-GBSA rescoring, we re-ranked docking hit lists in three small buried sites: a hydrophobic cavity that binds apolar ligands, a slightly polar cavity that binds aryl and hydrogen-bonding ligands, and an anionic cavity that binds cationic ligands. These sites are simple; consequently, incorrect predictions can be attributed to particular errors in the method, and many likely ligands may actually be tested. In retrospective calculations, MM-GBSA techniques with binding-site minimization better distinguished the known ligands for each cavity from the known decoys compared to the docking calculation alone. This encouraged us to test rescoring prospectively on molecules that ranked poorly by docking but that ranked well when rescored by MM-GBSA. A total of 33 molecules highly ranked by MM-GBSA for the three cavities were tested experimentally. Of these, 23 were observed to bind--these are docking false negatives rescued by rescoring. The 10 remaining molecules are true negatives by docking and false positives by MM-GBSA. X-ray crystal structures were determined for 21 of these 23 molecules. In many cases, the geometry prediction by MM-GBSA improved the initial docking pose and more closely resembled the crystallographic result; yet in several cases, the rescored geometry failed to capture large conformational changes in the protein. Intriguingly, rescoring not only rescued docking false positives, but also introduced several new false positives into the top-ranking molecules. We consider the origins of the successes and failures in MM-GBSA rescoring in these model cavity sites and the prospects for rescoring in biologically relevant targets.
Figures
Similar articles
-
Toward fully automated high performance computing drug discovery: a massively parallel virtual screening pipeline for docking and molecular mechanics/generalized Born surface area rescoring to improve enrichment.J Chem Inf Model. 2014 Jan 27;54(1):324-37. doi: 10.1021/ci4005145. Epub 2014 Jan 3. J Chem Inf Model. 2014. PMID: 24358939
-
A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin.Curr Top Med Chem. 2017;17(14):1631-1639. doi: 10.2174/1568026616666161117112604. Curr Top Med Chem. 2017. PMID: 27852201 Free PMC article.
-
Probing molecular docking in a charged model binding site.J Mol Biol. 2006 Apr 14;357(5):1449-70. doi: 10.1016/j.jmb.2006.01.034. Epub 2006 Feb 2. J Mol Biol. 2006. PMID: 16490206 Free PMC article.
-
Importance of molecular computer modeling in anticancer drug development.J BUON. 2007 Sep;12 Suppl 1:S101-18. J BUON. 2007. PMID: 17935268 Review.
-
The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities.Expert Opin Drug Discov. 2015 May;10(5):449-61. doi: 10.1517/17460441.2015.1032936. Epub 2015 Apr 2. Expert Opin Drug Discov. 2015. PMID: 25835573 Free PMC article. Review.
Cited by
-
Lipids Regulate Lck Protein Activity through Their Interactions with the Lck Src Homology 2 Domain.J Biol Chem. 2016 Aug 19;291(34):17639-50. doi: 10.1074/jbc.M116.720284. Epub 2016 Jun 22. J Biol Chem. 2016. PMID: 27334919 Free PMC article.
-
Three-Dimensional Modeling of Thyroid Hormone Metabolites Binding to the Cancer-Relevant αvβ3 Integrin: In-Silico Based Study.Front Endocrinol (Lausanne). 2022 May 27;13:895240. doi: 10.3389/fendo.2022.895240. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35692387 Free PMC article.
-
Precision therapeutic targeting of human cancer cell motility.Nat Commun. 2018 Jun 22;9(1):2454. doi: 10.1038/s41467-018-04465-5. Nat Commun. 2018. PMID: 29934502 Free PMC article.
-
Absolute Binding Free Energies between T4 Lysozyme and 141 Small Molecules: Calculations Based on Multiple Rigid Receptor Configurations.J Chem Theory Comput. 2017 Jun 13;13(6):2930-2944. doi: 10.1021/acs.jctc.6b01183. Epub 2017 May 1. J Chem Theory Comput. 2017. PMID: 28430432 Free PMC article.
-
Implicit ligand theory: rigorous binding free energies and thermodynamic expectations from molecular docking.J Chem Phys. 2012 Sep 14;137(10):104106. doi: 10.1063/1.4751284. J Chem Phys. 2012. PMID: 22979849 Free PMC article.
References
-
- Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure. 2002;10:1013–1023. - PubMed
-
- Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J. Med. Chem. 2004;47:5381–5392. - PubMed
-
- Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr Identification of nonphosphate-containing small molecular weight inhibitors of the tyrosine kinase p56 Lck SH2 domain via in silico screening against the pY+3 binding site. J. Med. Chem. 2004;47:3502–3511. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
