

Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss
- PMID: 18281279
- PMCID: PMC2447638
- DOI: 10.1074/jbc.M707659200
Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss
Abstract
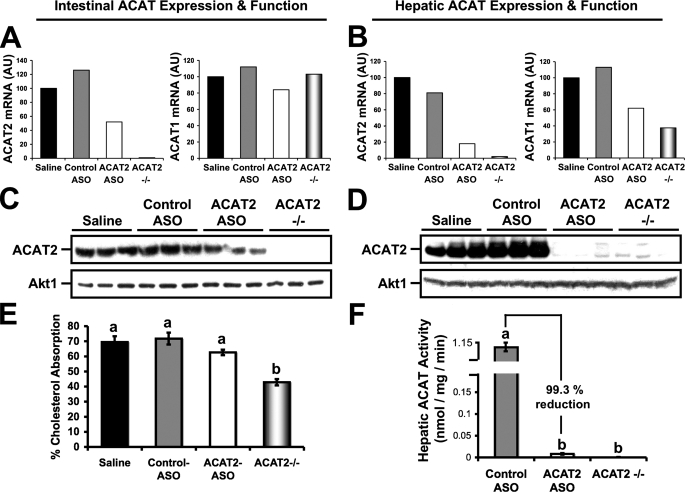
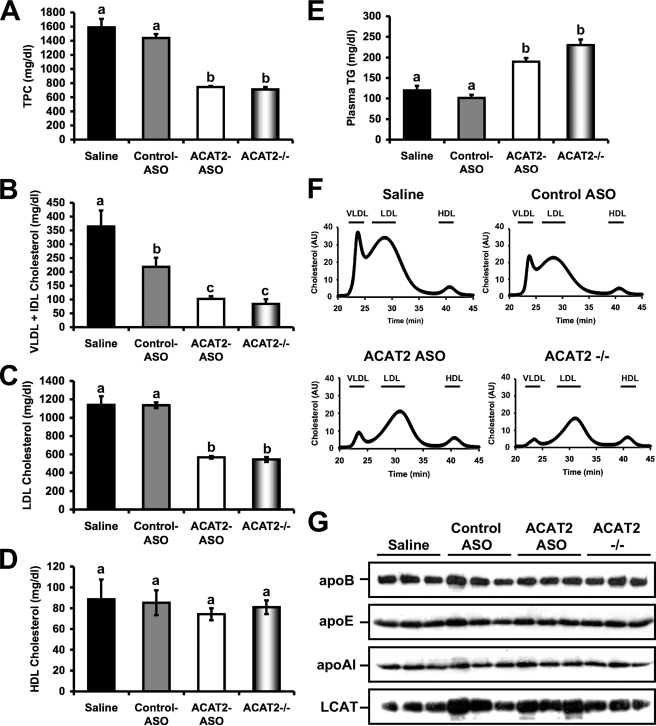
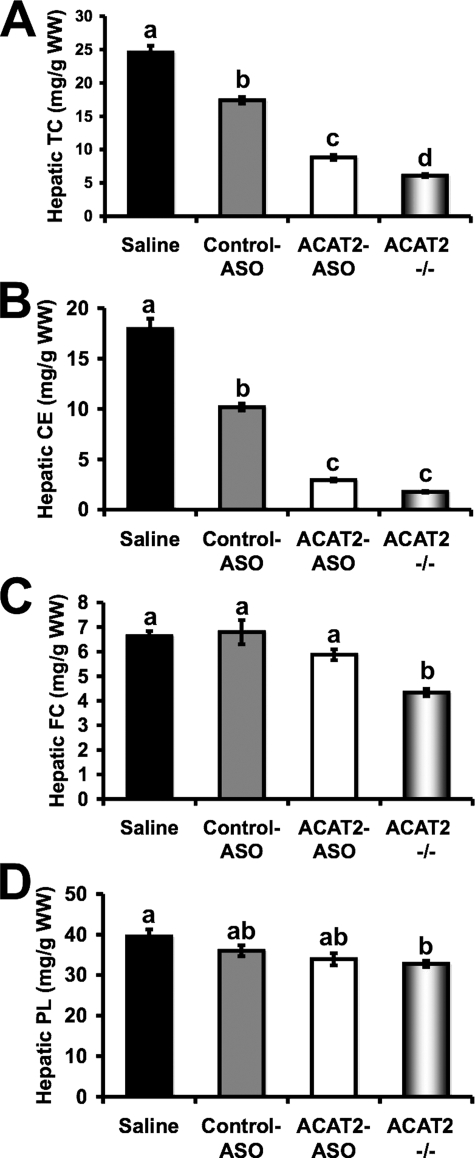
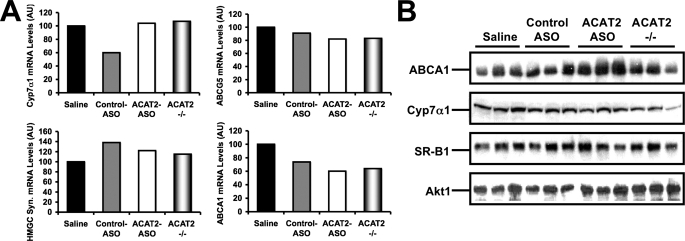
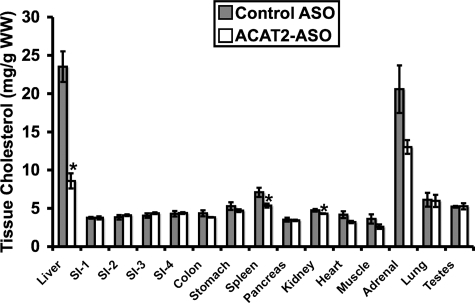
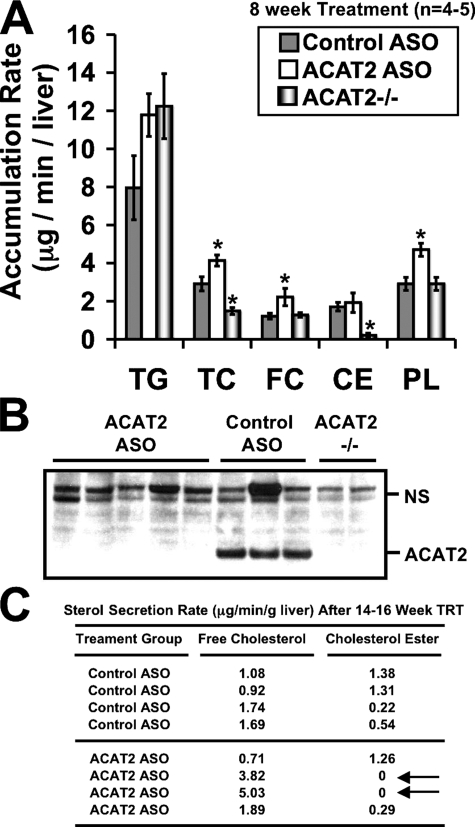
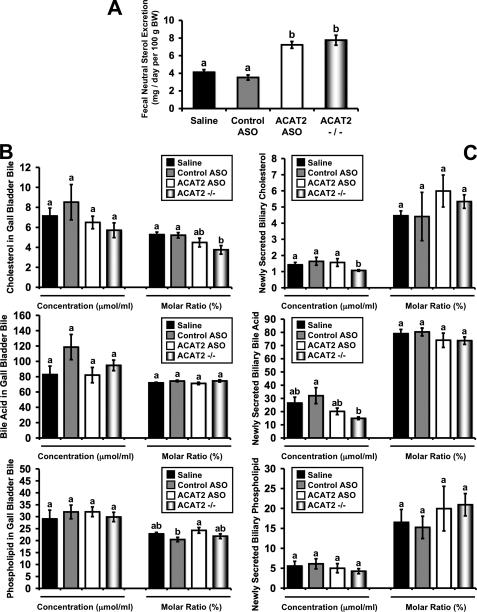
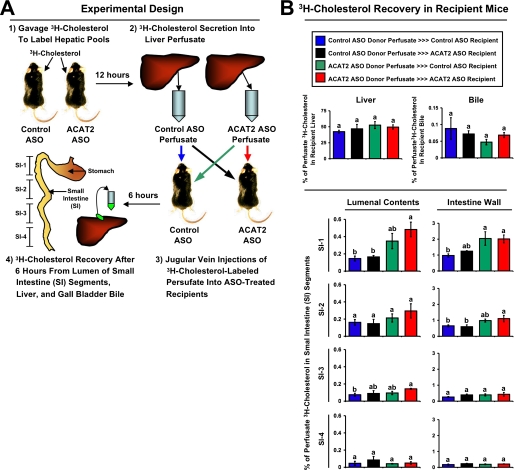
Deletion of acyl-CoA:cholesterol O-acyltransferase 2 (ACAT2) in mice results in resistance to diet-induced hypercholesterolemia and protection against atherosclerosis. Recently, our group has shown that liver-specific inhibition of ACAT2 via antisense oligonucleotide (ASO)-mediated targeting likewise limits atherosclerosis. However, whether this atheroprotective effect was mediated by: 1) prevention of packaging of cholesterol into apoB-containing lipoproteins, 2) augmentation of nascent HDL cholesterol secretion, or 3) increased hepatobiliary sterol secretion was not examined. Therefore, the purpose of these studies was to determine whether hepatic ACAT2 is rate-limiting in all three of these important routes of cholesterol homeostasis. Liver-specific depletion of ACAT2 resulted in reduced packaging of cholesterol into apoB-containing lipoproteins (very low density lipoprotein, intermediate density lipoprotein, and low density lipoprotein), whereas high density lipoprotein cholesterol levels remained unchanged. In the liver of ACAT2 ASO-treated mice, cholesterol ester accumulation was dramatically reduced, yet there was no reciprocal accumulation of unesterified cholesterol. Paradoxically, ASO-mediated depletion of hepatic ACAT2 promoted fecal neutral sterol excretion without altering biliary sterol secretion. Interestingly, during isolated liver perfusion, ACAT2 ASO-treated livers had augmented secretion rates of unesterified cholesterol and phospholipid. Furthermore, we demonstrate that liver-derived cholesterol from ACAT2 ASO-treated mice is preferentially delivered to the proximal small intestine as a precursor to fecal excretion. Collectively, these studies provide the first insight into the hepatic itinerary of cholesterol when cholesterol esterification is inhibited only in the liver, and provide evidence for a novel non-biliary route of fecal sterol loss.
Figures
Comment in
-
A new twist in the cholesterol metabolism-based enterohepatic connection.Gastroenterology. 2008 Aug;135(2):705-6. doi: 10.1053/j.gastro.2008.06.067. Epub 2008 Jul 9. Gastroenterology. 2008. PMID: 18619455 No abstract available.
References
-
- Dietschy, J. M., and Turley, S. D. (2002) J. Biol. Chem. 277 3801-3804 - PubMed
-
- Dietschy, J. M., Turley, S. D., and Spady, D. K. (1993) J. Lipid Res. 34 1637-1659 - PubMed
-
- Brown, M. S., Kovanen, P. T., and Goldstein, J. L. (1981) Science 212 628-635 - PubMed
-
- Turley, S. D., and Dietshcy, J. M. (2003) Curr. Opin. Lipidol. 14 233-240 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
