

Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells
- PMID: 18285502
- PMCID: PMC2279248
- DOI: 10.1101/gr.7179508
Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells
Erratum in
- Genome Res. 2009 May;19(5):958
Abstract
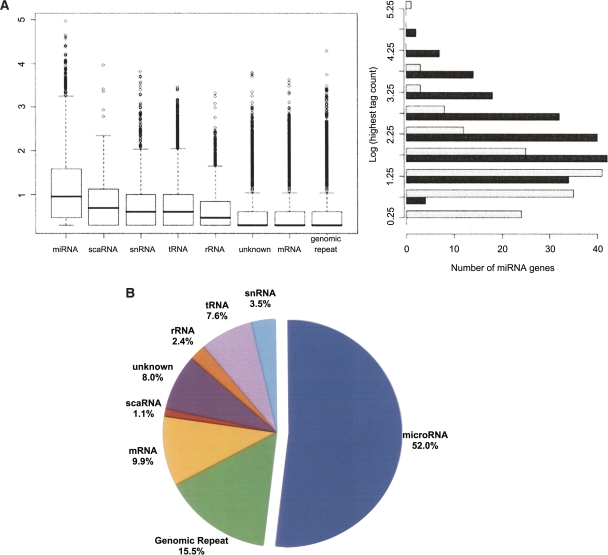
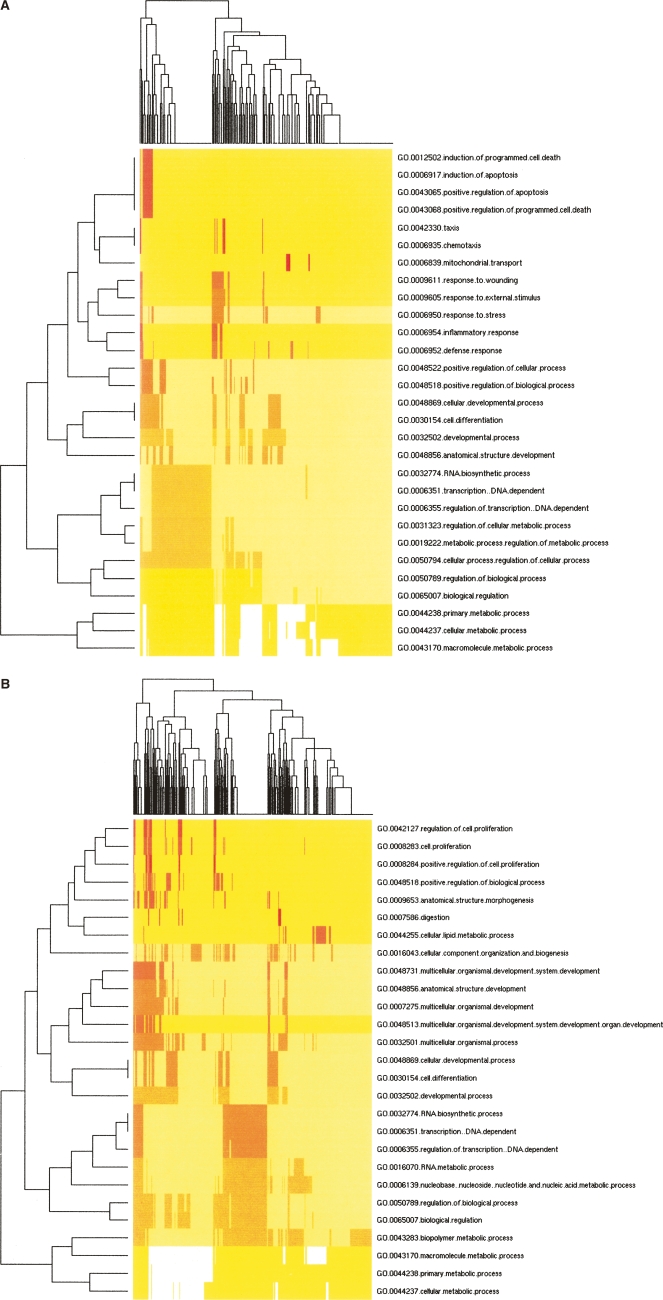
MicroRNAs (miRNAs) are emerging as important, albeit poorly characterized, regulators of biological processes. Key to further elucidation of their roles is the generation of more complete lists of their numbers and expression changes in different cell states. Here, we report a new method for surveying the expression of small RNAs, including microRNAs, using Illumina sequencing technology. We also present a set of methods for annotating sequences deriving from known miRNAs, identifying variability in mature miRNA sequences, and identifying sequences belonging to previously unidentified miRNA genes. Application of this approach to RNA from human embryonic stem cells obtained before and after their differentiation into embryoid bodies revealed the sequences and expression levels of 334 known plus 104 novel miRNA genes. One hundred seventy-one known and 23 novel microRNA sequences exhibited significant expression differences between these two developmental states. Owing to the increased number of sequence reads, these libraries represent the deepest miRNA sampling to date, spanning nearly six orders of magnitude of expression. The predicted targets of those miRNAs enriched in either sample shared common features. Included among the high-ranked predicted gene targets are those implicated in differentiation, cell cycle control, programmed cell death, and transcriptional regulation.
Figures

References
-
- Abeyta M.J., Clark A.T., Rodriguez R.T., Bodnar M.S., Pera R.A., Firpo M.T., Clark A.T., Rodriguez R.T., Bodnar M.S., Pera R.A., Firpo M.T., Rodriguez R.T., Bodnar M.S., Pera R.A., Firpo M.T., Bodnar M.S., Pera R.A., Firpo M.T., Pera R.A., Firpo M.T., Firpo M.T. Unique gene expression signatures of independently-derived human embryonic stem cell lines. Hum. Mol. Genet. 2004;13:601–608. - PubMed
-
- Aravin A., Tuschl T., Tuschl T. Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett. 2005;579:5830–5840. - PubMed
-
- Aravin A.A., Sachidanandam R., Girard A., Fejes-Toth K., Hannon G.J., Sachidanandam R., Girard A., Fejes-Toth K., Hannon G.J., Girard A., Fejes-Toth K., Hannon G.J., Fejes-Toth K., Hannon G.J., Hannon G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316:744–747. - PubMed
-
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Dolinski K., Dwight S.S., Eppig J.T., Dwight S.S., Eppig J.T., Eppig J.T., et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. - PMC - PubMed
-
- Audic S., Claverie J.M., Claverie J.M. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources