

Chemical rescue of deltaF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells
- PMID: 18285607
- PMCID: PMC2424193
- DOI: 10.1074/mcp.M700303-MCP200
Chemical rescue of deltaF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells
Abstract
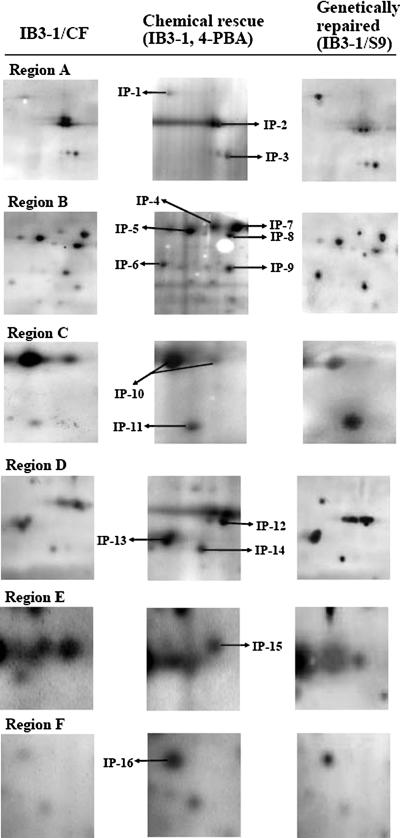
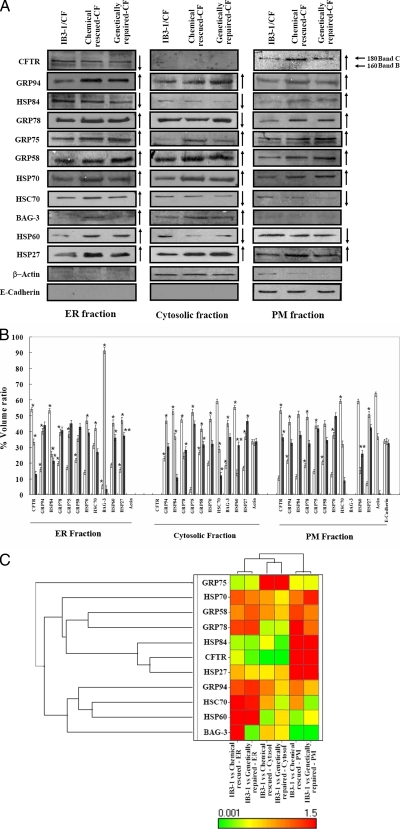
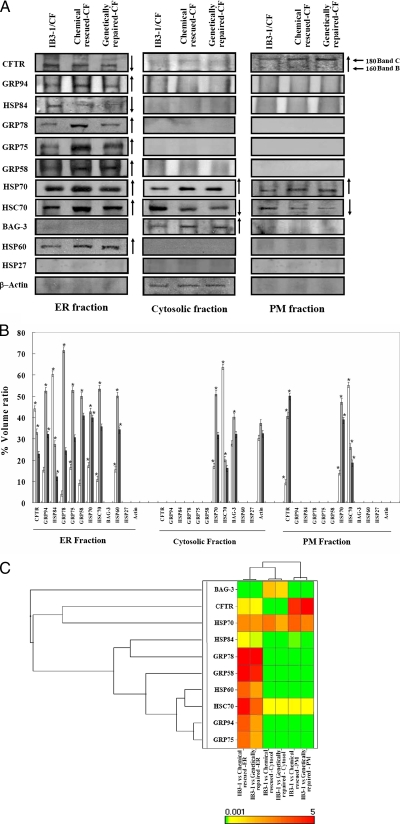
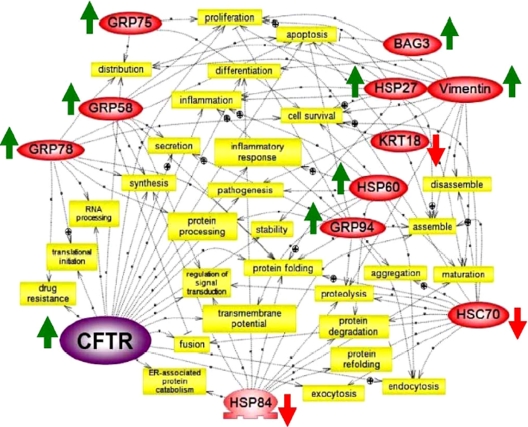
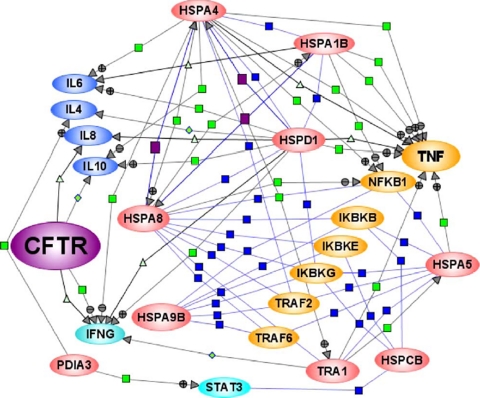
In a previous study of sodium 4-phenylbutyrate (4-PBA)-responsive proteins in cystic fibrosis (CF) IB3-1 bronchial epithelial cells, we identified 85 differentially expressed high abundance proteins from whole cellular lysate (Singh, O. V., Vij, N., Mogayzel, P. J., Jr., Jozwik, C., Pollard, H. B., and Zeitlin, P. L. (2006) Pharmacoproteomics of 4-phenylbutyrate-treated IB3-1 cystic fibrosis bronchial epithelial cells. J. Proteome Res. 5, 562-571). In the present work we hypothesize that a subset of heat shock proteins that interact with cystic fibrosis transmembrane conductance regulator (CFTR) in common during chemical rescue and genetic repair will identify therapeutic networks for targeted intervention. Immunocomplexes were generated from total cellular lysates, and three subcellular fractions (endoplasmic reticulum (ER), cytosol, and plasma membrane) with anti-CFTR polyclonal antibody from CF (IB3-1), chemically rescued CF (4-PBA-treated IB3-1), and genetically repaired CF (IB3-1/S9 daughter cells repaired by gene transfer with adeno-associated virus-(wild type) CFTR). CFTR-interacting proteins were analyzed on two-dimensional gels and identified by mass spectrometry. A set of 16 proteins known to act in ER-associated degradation were regulated in common and functionally connected to the protein processing, protein folding, and inflammatory response. Some of these proteins were modulated exclusively in ER, cytosol, or plasma membrane. A subset of 4-PBA-modulated ER-associated degradation chaperones (GRP94, HSP84, GRP78, GRP75, and GRP58) was observed to associate with the immature B form of CFTR in ER. HSP70 and HSC70 interacted with the C band (mature form) of CFTR at the cell surface. We conclude that chemically rescued CFTR associates with a specific set of HSP70 family proteins that mark therapeutic interactions and can be useful to correct both ion transport and inflammatory phenotypes in CF subjects.
Figures

References
-
- Tsui, L. C. ( 1992) Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium. Hum. Mutat. 1, 197–203 - PubMed
-
- Ward, C. L., Omura, S., and Kopito, R. R. ( 1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 - PubMed
-
- Gabriel, S. E., Clarke, L. L., Boucher, R. C., and Stutts, M. J. ( 1993) CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature 363, 263–268 - PubMed
-
- Stutts, M. J., Canessa, C. M., Olsen, J. C., Hamrick, M., Cohn, J. A., Rossier, B. C., and Boucher, R. C. ( 1995) CFTR as a cAMP-dependent regulator of sodium channels. Science 269, 847–850 - PubMed
-
- Amaral, M. D. ( 2004) CFTR and chaperones: processing and degradation. J. Mol. Neurosci. 23, 41–48 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
