

Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick
- PMID: 18286181
- PMCID: PMC2230226
- DOI: 10.1371/journal.pone.0001624
Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick
Abstract
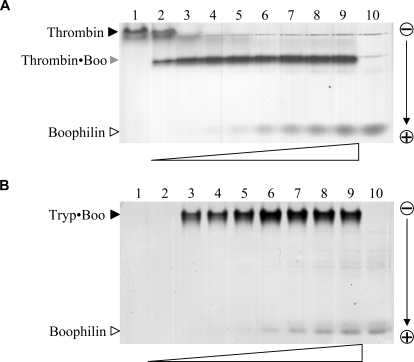
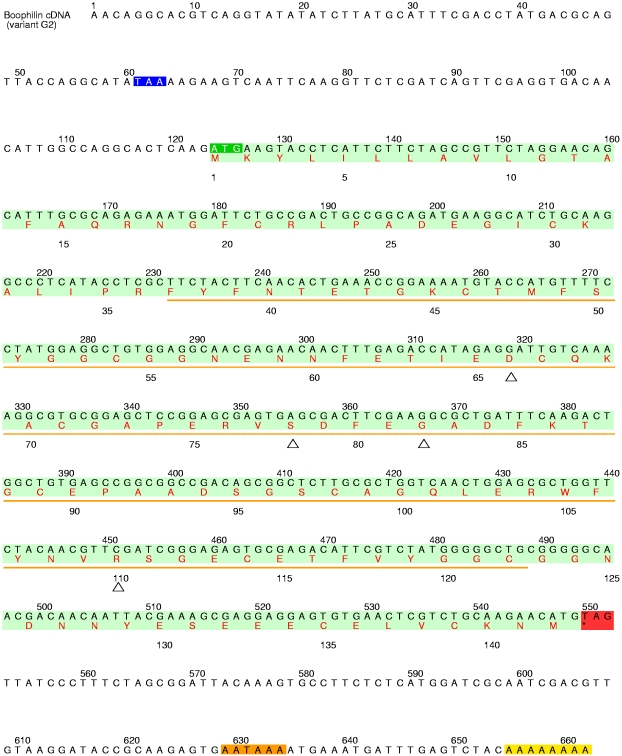
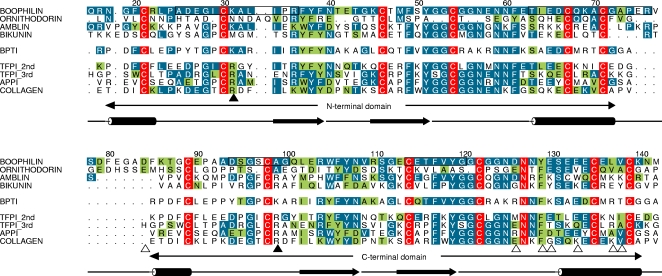
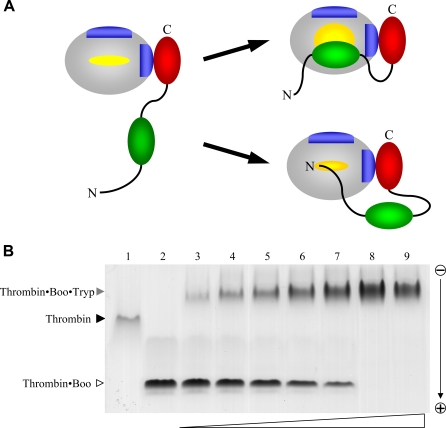
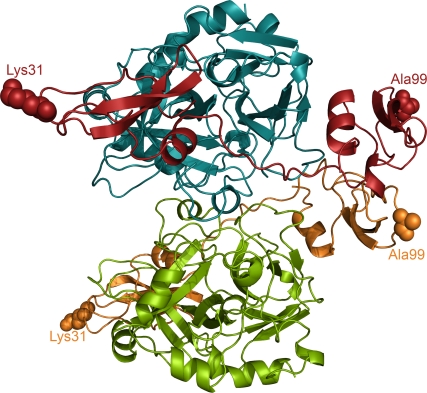
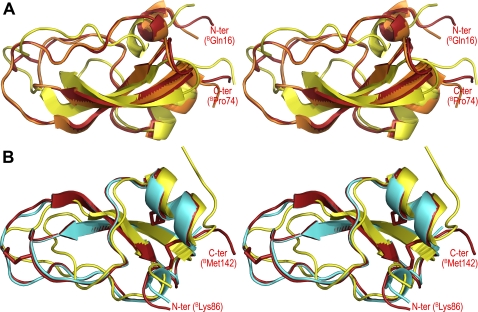
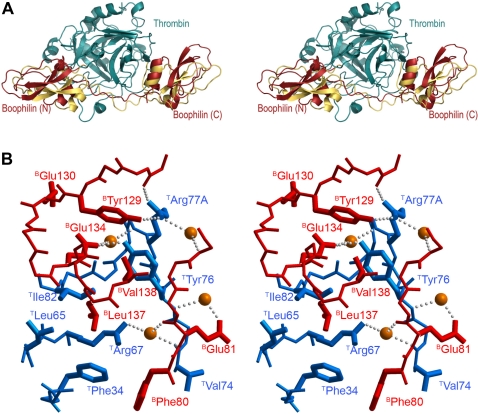
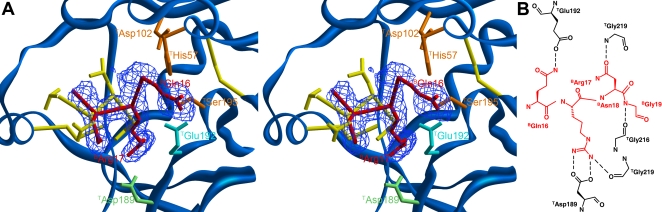
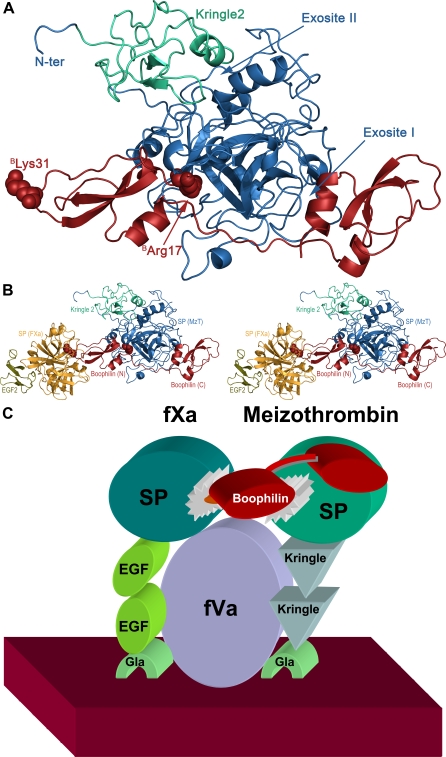
Inhibitors of coagulation factors from blood-feeding animals display a wide variety of structural motifs and inhibition mechanisms. We have isolated a novel inhibitor from the cattle tick Boophilus microplus, one of the most widespread parasites of farm animals. The inhibitor, which we have termed boophilin, has been cloned and overexpressed in Escherichia coli. Mature boophilin is composed of two canonical Kunitz-type domains, and inhibits not only the major procoagulant enzyme, thrombin, but in addition, and by contrast to all other previously characterised natural thrombin inhibitors, significantly interferes with the proteolytic activity of other serine proteinases such as trypsin and plasmin. The crystal structure of the bovine alpha-thrombin.boophilin complex, refined at 2.35 A resolution reveals a non-canonical binding mode to the proteinase. The N-terminal region of the mature inhibitor, Q16-R17-N18, binds in a parallel manner across the active site of the proteinase, with the guanidinium group of R17 anchored in the S(1) pocket, while the C-terminal Kunitz domain is negatively charged and docks into the basic exosite I of thrombin. This binding mode resembles the previously characterised thrombin inhibitor, ornithodorin which, unlike boophilin, is composed of two distorted Kunitz modules. Unexpectedly, both boophilin domains adopt markedly different orientations when compared to those of ornithodorin, in its complex with thrombin. The N-terminal boophilin domain rotates 9 degrees and is displaced by 6 A, while the C-terminal domain rotates almost 6 degrees accompanied by a 3 A displacement. The reactive-site loop of the N-terminal Kunitz domain of boophilin with its P(1) residue, K31, is fully solvent exposed and could thus bind a second trypsin-like proteinase without sterical restraints. This finding explains the formation of a ternary thrombin.boophilin.trypsin complex, and suggests a mechanism for prothrombinase inhibition in vivo.
Conflict of interest statement
Figures
Similar articles
-
In Vitro Mode of Action and Anti-thrombotic Activity of Boophilin, a Multifunctional Kunitz Protease Inhibitor from the Midgut of a Tick Vector of Babesiosis, Rhipicephalus microplus.PLoS Negl Trop Dis. 2016 Jan 8;10(1):e0004298. doi: 10.1371/journal.pntd.0004298. eCollection 2016 Jan. PLoS Negl Trop Dis. 2016. PMID: 26745503 Free PMC article.
-
Tick-derived Kunitz-type inhibitors as antihemostatic factors.Insect Biochem Mol Biol. 2009 Sep;39(9):579-95. doi: 10.1016/j.ibmb.2009.07.003. Epub 2009 Jul 24. Insect Biochem Mol Biol. 2009. PMID: 19631744 Review.
-
Expression and functional characterization of boophilin, a thrombin inhibitor from Rhipicephalus (Boophilus) microplus midgut.Vet Parasitol. 2012 Jul 6;187(3-4):521-8. doi: 10.1016/j.vetpar.2012.01.027. Epub 2012 Jan 28. Vet Parasitol. 2012. PMID: 22341830
-
Crystallization and preliminary crystallographic characterization of the N-terminal Kunitz domain of boophilin.Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012 Apr 1;68(Pt 4):436-9. doi: 10.1107/S1744309112005532. Epub 2012 Mar 27. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012. PMID: 22505414 Free PMC article.
-
[New studies on natural inhibitors of proteolytic enzymes].Bioorg Khim. 1998 May;24(5):332-40. Bioorg Khim. 1998. PMID: 9661786 Review. Russian.
Cited by
-
Evolution, expansion and expression of the Kunitz/BPTI gene family associated with long-term blood feeding in Ixodes Scapularis.BMC Evol Biol. 2012 Jan 14;12:4. doi: 10.1186/1471-2148-12-4. BMC Evol Biol. 2012. PMID: 22244187 Free PMC article.
-
Characterization of Ixophilin, a thrombin inhibitor from the gut of Ixodes scapularis.PLoS One. 2013 Jul 9;8(7):e68012. doi: 10.1371/journal.pone.0068012. Print 2013. PLoS One. 2013. PMID: 23874485 Free PMC article.
-
Silencing of a putative immunophilin gene in the cattle tick Rhipicephalus (Boophilus) microplus increases the infection rate of Babesia bovis in larval progeny.Parasit Vectors. 2009 Nov 20;2(1):57. doi: 10.1186/1756-3305-2-57. Parasit Vectors. 2009. PMID: 19930572 Free PMC article.
-
Comparative microarray analysis of Rhipicephalus (Boophilus) microplus expression profiles of larvae pre-attachment and feeding adult female stages on Bos indicus and Bos taurus cattle.BMC Genomics. 2010 Jul 19;11:437. doi: 10.1186/1471-2164-11-437. BMC Genomics. 2010. PMID: 20637126 Free PMC article.
-
In Vitro Mode of Action and Anti-thrombotic Activity of Boophilin, a Multifunctional Kunitz Protease Inhibitor from the Midgut of a Tick Vector of Babesiosis, Rhipicephalus microplus.PLoS Negl Trop Dis. 2016 Jan 8;10(1):e0004298. doi: 10.1371/journal.pntd.0004298. eCollection 2016 Jan. PLoS Negl Trop Dis. 2016. PMID: 26745503 Free PMC article.
References
-
- Bork P, Downing AK, Kieffer B, Campbell ID. Structure and distribution of modules in extracellular proteins. Q Rev Biophys. 1996;29:119–167. - PubMed
-
- Laskowski M, Jr, Kato I. Protein inhibitors of proteinases. Annu Rev Biochem. 1980;49:593–626. - PubMed
-
- Ascenzi P, Bocedi A, Bolognesi M, Spallarossa A, Coletta M, et al. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Curr Protein Pept Sci. 2003;4:231–251. - PubMed
-
- Broze GJ., Jr Tissue factor pathway inhibitor and the revised theory of coagulation. Annu Rev Med. 1995;46:103–112. - PubMed
-
- Huber R, Kukla D, Bode W, Schwager P, Bartels K, et al. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. II. Crystallographic refinement at 1.9 Å resolution. J Mol Biol. 1974;89:73–101. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
