

Requirement of protein kinase D1 for pathological cardiac remodeling
- PMID: 18287012
- PMCID: PMC2268584
- DOI: 10.1073/pnas.0712265105
Requirement of protein kinase D1 for pathological cardiac remodeling
Abstract
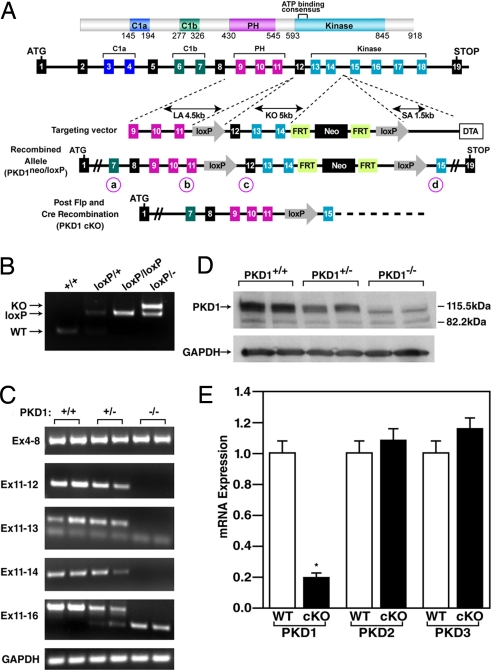
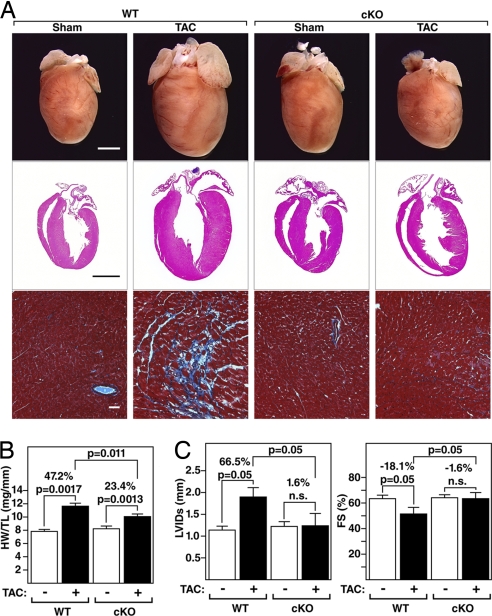
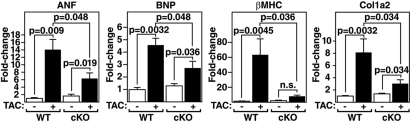
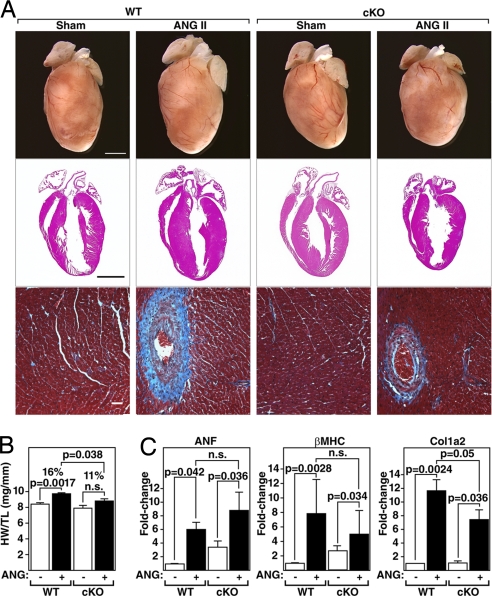
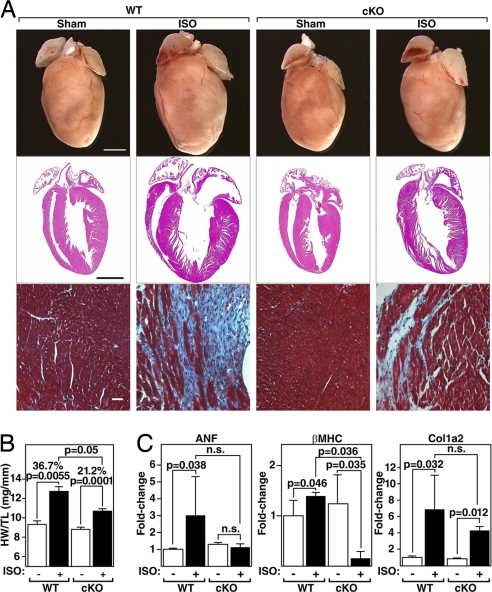
The adult heart responds to biomechanical stress and neurohormonal signaling by hypertrophic growth, accompanied by fibrosis, diminished pump function, and activation of a fetal gene program. Class II histone deacetylases (HDACs) suppress stress-dependent remodeling of the heart via their association with the MEF2 transcription factor, an activator of heart disease. Protein kinase D (PKD) is a stress-responsive kinase that phosphorylates class II HDACs, resulting in their dissociation from MEF2 with consequent activation of MEF2 target genes. To test whether PKD1 is required for pathological cardiac remodeling in vivo, we generated mice with a conditional PKD1-null allele. Mice with cardiac-specific deletion of PKD1 were viable and showed diminished hypertrophy, fibrosis, and fetal gene activation as well as improved cardiac function in response to pressure overload or chronic adrenergic and angiotensin II signaling. We conclude that PKD1 functions as a key transducer of stress stimuli involved in pathological cardiac remodeling in vivo.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. - PubMed
-
- Marks AR. A guide for the perplexed: Towards an understanding of the molecular basis of heart failure. Circulation. 2003;107:1456–1459. - PubMed
-
- Olson EN, Schneider MD. Sizing up the heart: Development redux in disease. Genes Dev. 2003;17:1937–1956. - PubMed
-
- Gardin JM, Lauer MS. Left ventricular hypertrophy: The next treatable, silent killer? J Am Med Assoc. 2004;292:2396–2398. - PubMed
-
- Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
