

Elevated globotriaosylsphingosine is a hallmark of Fabry disease
- PMID: 18287059
- PMCID: PMC2268542
- DOI: 10.1073/pnas.0712309105
Elevated globotriaosylsphingosine is a hallmark of Fabry disease
Abstract
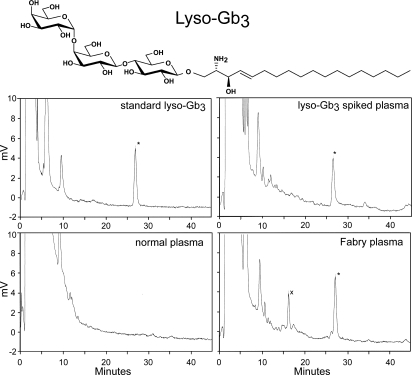
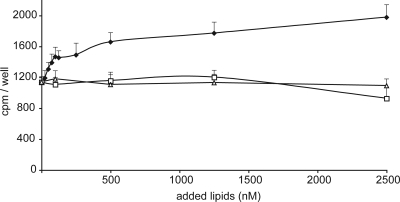
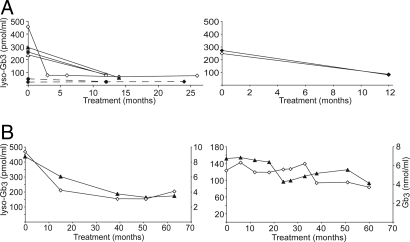
Fabry disease is an X-linked lysosomal storage disease caused by deficiency of alpha-galactosidase A that affects males and shows disease expression in heterozygotes. The characteristic progressive renal insufficiency, cardiac involvement, and neuropathology usually are ascribed to globotriaosylceramide accumulation in the endothelium. However, no direct correlation exists between lipid storage and clinical manifestations, and treatment of patients with recombinant enzymes does not reverse several key signs despite clearance of lipid from the endothelium. We therefore investigated the possibility that globotriaosylceramide metabolites are a missing link in the pathogenesis. We report that deacylated globotriaosylceramide, globotriaosylsphingosine, and a minor additional metabolite are dramatically increased in plasma of classically affected male Fabry patients and plasma and tissues of Fabry mice. Plasma globotriaosylceramide levels are reduced by therapy. We show that globotriaosylsphingosine is an inhibitor of alpha-galactosidase A activity. Furthermore, exposure of smooth muscle cells, but not fibroblasts, to globotriaosylsphingosine at concentrations observed in plasma of patients promotes proliferation. The increased intima-media thickness in Fabry patients therefore may be related to the presence of this metabolite. Our findings suggest that measurement of circulating globotriaosylsphingosine will be useful to monitor Fabry disease and may contribute to a better understanding of the disorder.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Brady RO, et al. Enzymatic defect in Fabry's disease: Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163–1167. - PubMed
-
- Kint JA. Fabry's disease: α-Galactosidase deficiency. Science. 1970;167:1268–1269. - PubMed
-
- Desnick RJ, Ioannou YA. α-Galactosidase A deficiency: Fabry disease. In: Scriver R, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3733–3774.
-
- Brady RO, et al. Replacement therapy for inherited enzyme deficiency: Use of purified ceramidetrihexosidase in Fabry's disease. N Engl J Med. 1973;289:9–14. - PubMed
-
- Schiffmann R, et al. Enzyme-replacement therapy in Fabry disease: A randomized controlled trial. J Am Med Assoc. 2001;285:2743–2749. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
