

Dipeptidyl peptidase I-dependent neutrophil recruitment modulates the inflammatory response to Sendai virus infection
- PMID: 18292580
- PMCID: PMC2597084
- DOI: 10.4049/jimmunol.180.5.3535
Dipeptidyl peptidase I-dependent neutrophil recruitment modulates the inflammatory response to Sendai virus infection
Abstract
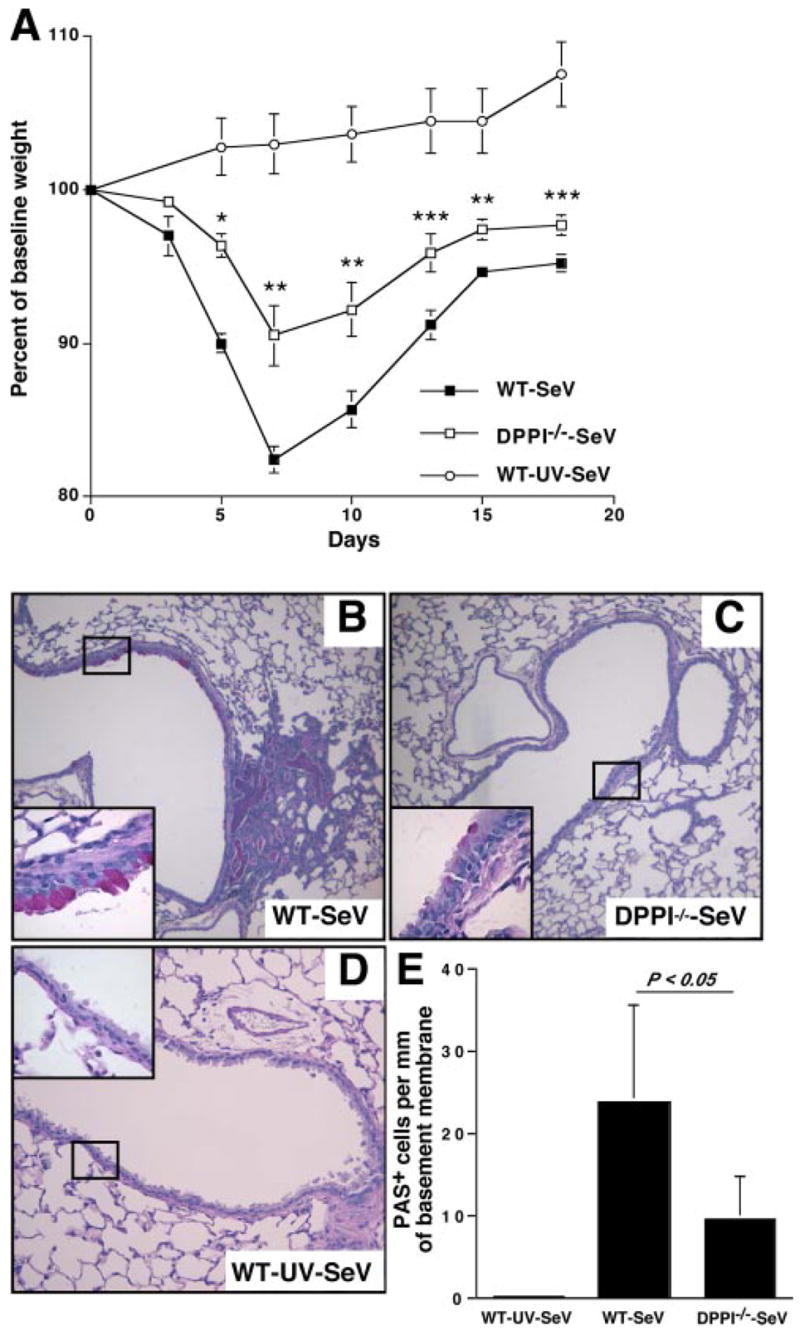
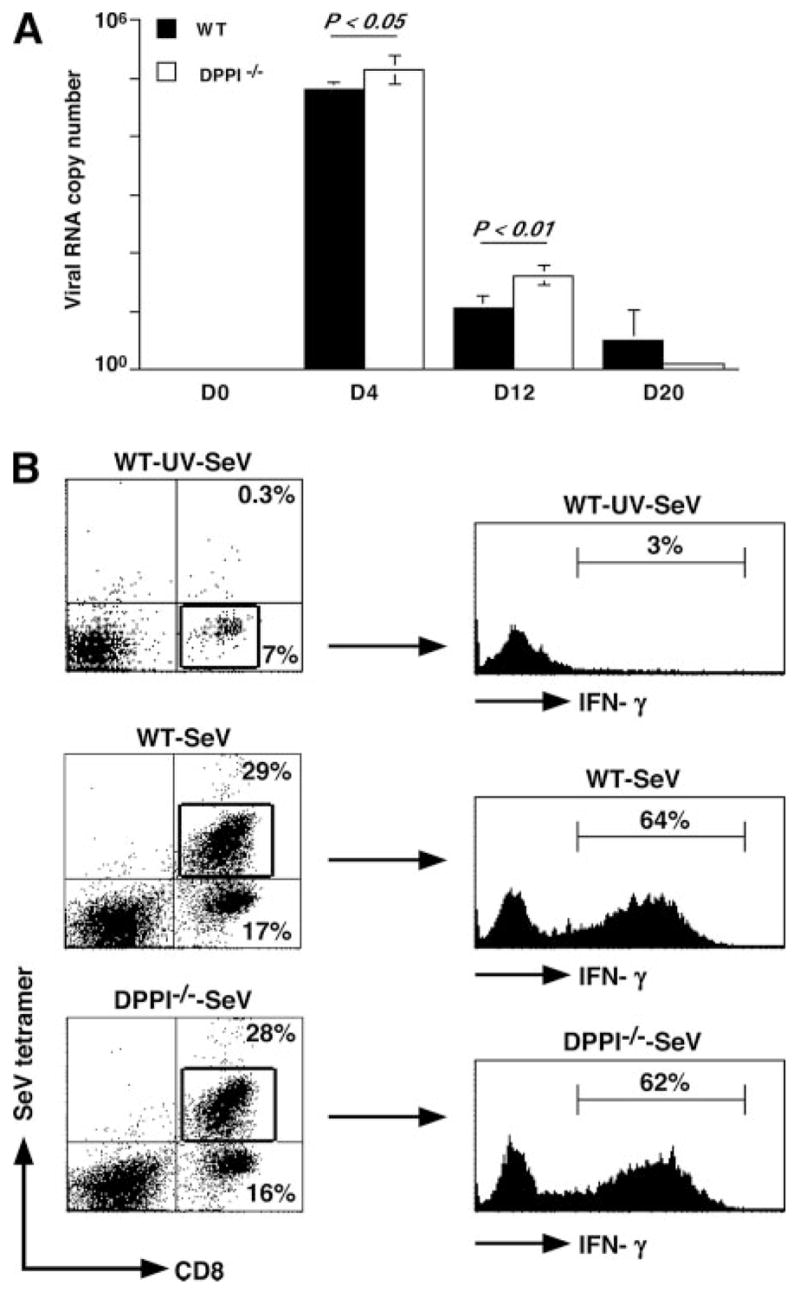
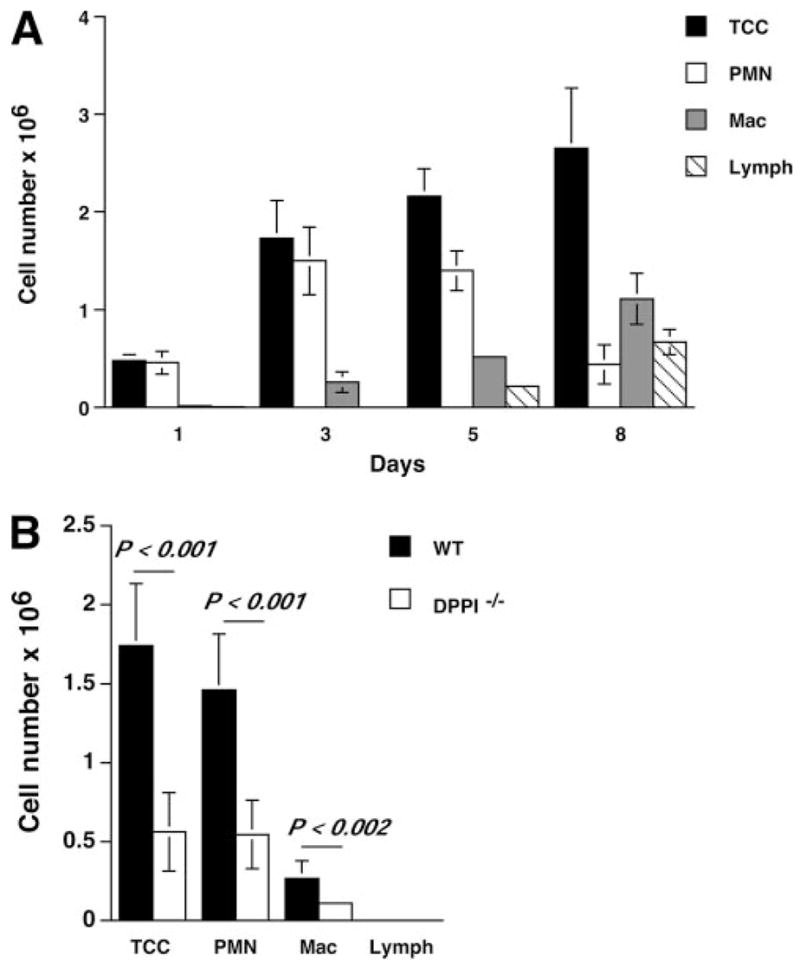
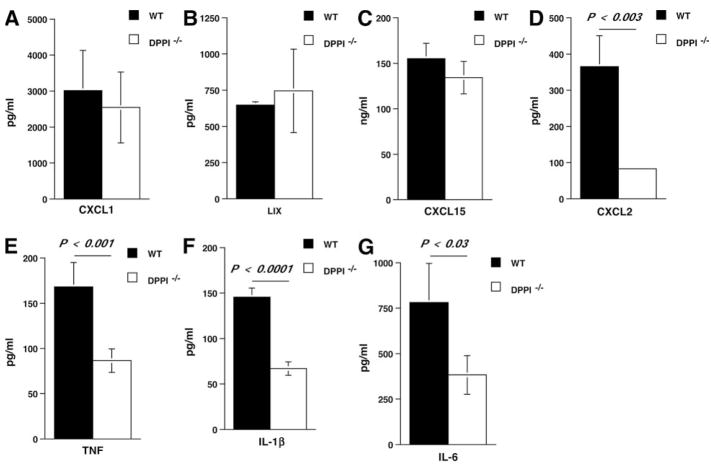
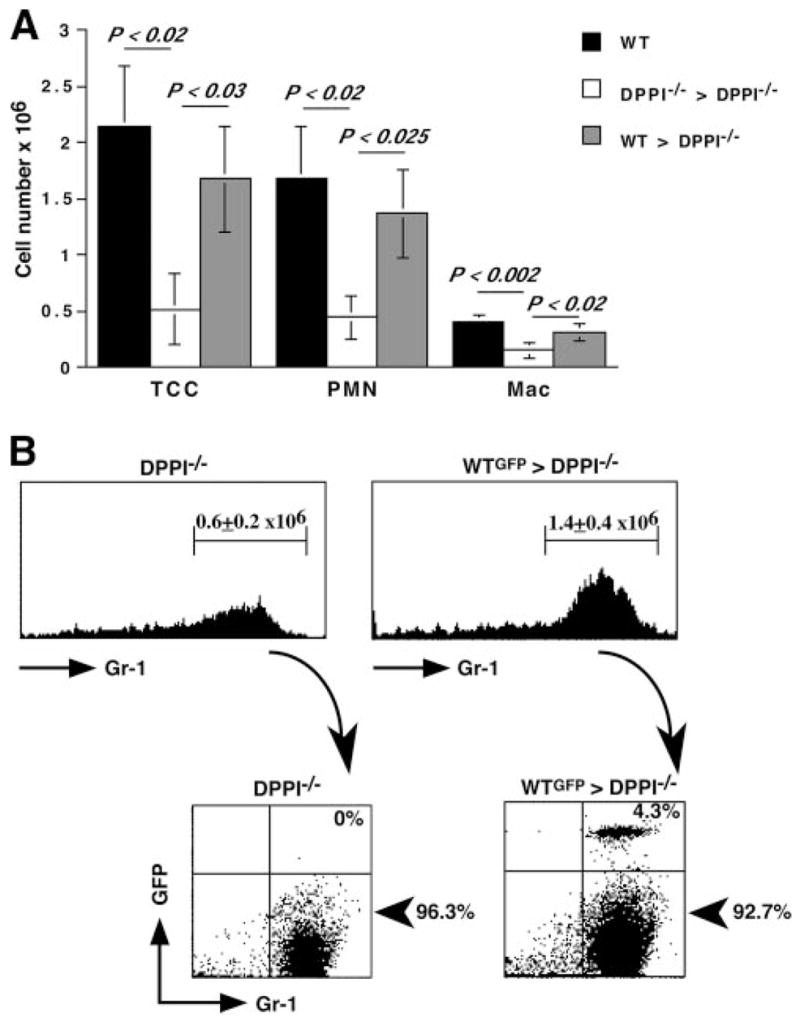
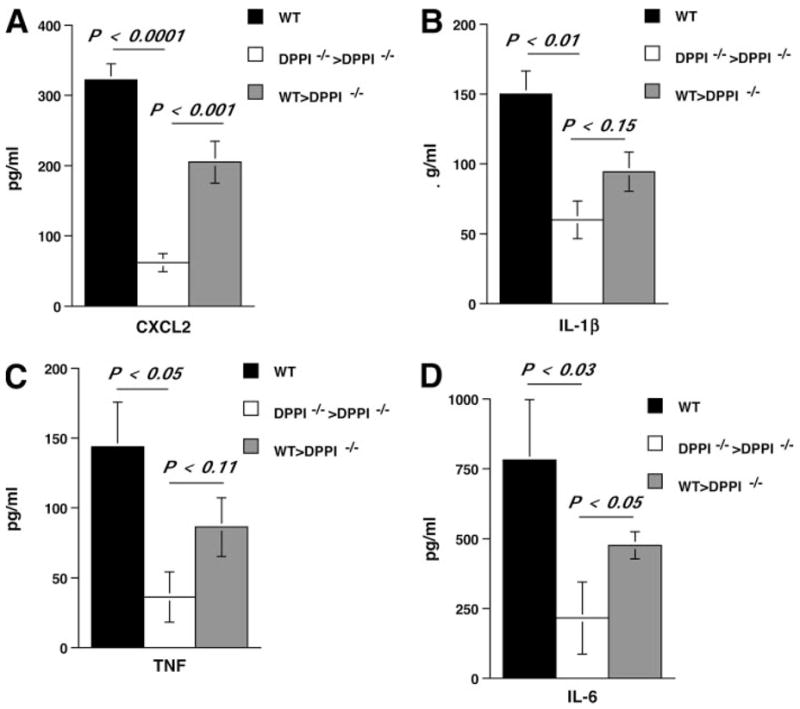
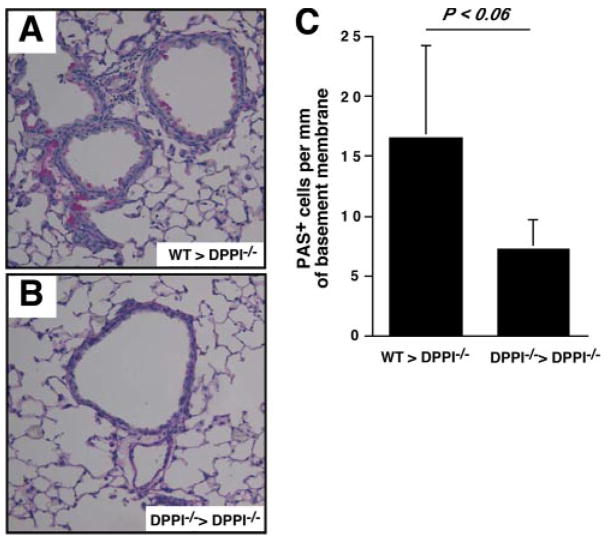
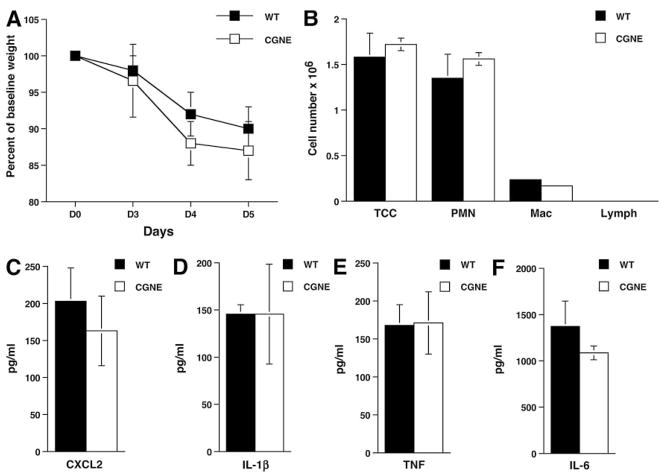
The role of innate immunity in the pathogenesis of asthma is unclear. Although increased presence of neutrophils is associated with persistent asthma and asthma exacerbations, how neutrophils participate in the pathogenesis of asthma remains controversial. In this study, we show that the absence of dipeptidyl peptidase I (DPPI), a lysosomal cysteine protease found in neutrophils, dampens the acute inflammatory response and the subsequent mucous cell metaplasia that accompanies the asthma phenotype induced by Sendai virus infection. This attenuated phenotype is accompanied by a significant decrease in the accumulation of neutrophils and the local production of CXCL2, TNF, IL-1beta, and IL-6 in the lung of infected DPPI-/- mice. Adoptive transfer of DPPI-sufficient neutrophils into DPPI-/- mice restored the levels of CXCL2 and enhanced cytokine production on day 4 postinfection and subsequent mucous cell metaplasia on day 21 postinfection. These results indicate that DPPI and neutrophils play a critical role in Sendai virus-induced asthma phenotype as a result of a DPPI-dependent neutrophil recruitment and cytokine response.
Figures
References
-
- Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. - PubMed
-
- Humbert M, Durham SR, Ying S, Kimmitt P, Barkans J, Assoufi B, Pfister R, Menz G, Robinson DS, Kay AB, Corrigan CJ. IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients with atopic and nonatopic asthma: evidence against “intrinsic” asthma being a distinct immunopathologic entity. Am J Respir Crit Care Med. 1996;154:1497–1504. - PubMed
-
- Temann UA, Prasad B, Gallup MW, Basbaum C, Ho SB, Flavell RA, Rankin JA. A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion. Am J Respir Cell Mol Biol. 1997;16:471–478. - PubMed
-
- Webb DC, McKenzie AN, Koskinen AM, Yang M, Mattes J, Foster PS. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J Immunol. 2000;165:108–113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
