

Bisulfite sequencing Data Presentation and Compilation (BDPC) web server--a useful tool for DNA methylation analysis
- PMID: 18296484
- PMCID: PMC2275153
- DOI: 10.1093/nar/gkn083
Bisulfite sequencing Data Presentation and Compilation (BDPC) web server--a useful tool for DNA methylation analysis
Abstract
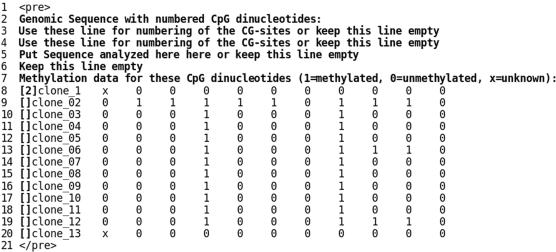
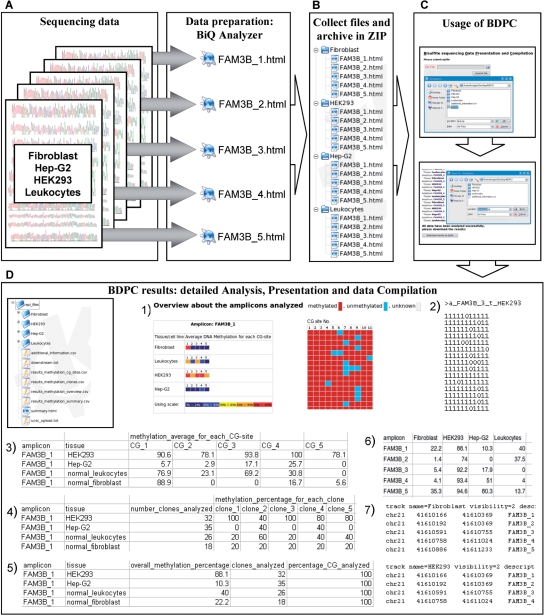
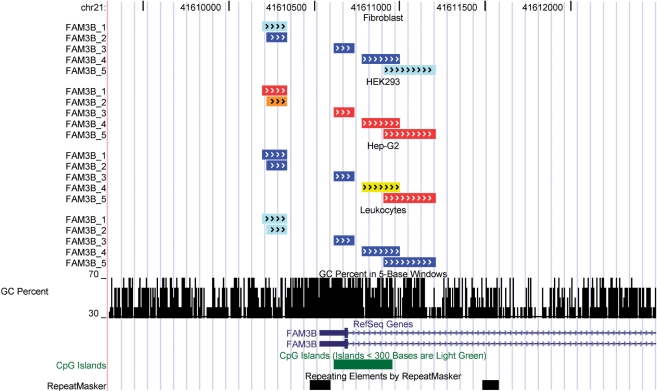
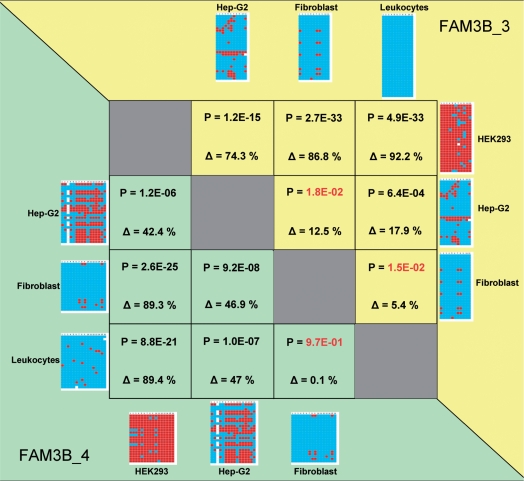
During bisulfite genomic sequencing projects large amount of data are generated. The Bisulfite sequencing Data Presentation and Compilation (BDPC) web interface (http://biochem.jacobs-university.de/BDPC/) automatically analyzes bisulfite datasets prepared using the BiQ Analyzer. BDPC provides the following output: (i) MS-Excel compatible files compiling for each PCR product (a) the average methylation level, the number of clones analyzed and the percentage of CG sites analyzed (which is an indicator of data quality), (b) the methylation level observed at each CG site and (c) the methylation level of each clone. (ii) A methylation overview table compiling the methylation of all amplicons in all tissues. (iii) Publication grade figures in PNG format showing the methylation pattern for each PCR product embedded in an HMTL file summarizing the methylation data, the DNA sequence and some basic statistics. (iv) A summary file compiling the methylation pattern of different tissues, which is linked to the individual HTML result files, and can be directly used for presentation of the data in the internet. (v) A condensed file, containing all primary data in simplified format for further downstream data analysis and (vi) a custom track file for display of the results in the UCSC genome browser.
Figures


References
-
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. - PubMed
-
- Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr. Opin. Cell Biol. 2007;19:266–272. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
