

Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members
- PMID: 18297114
- PMCID: PMC2903615
- DOI: 10.1038/onc.2008.19
Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members
Abstract
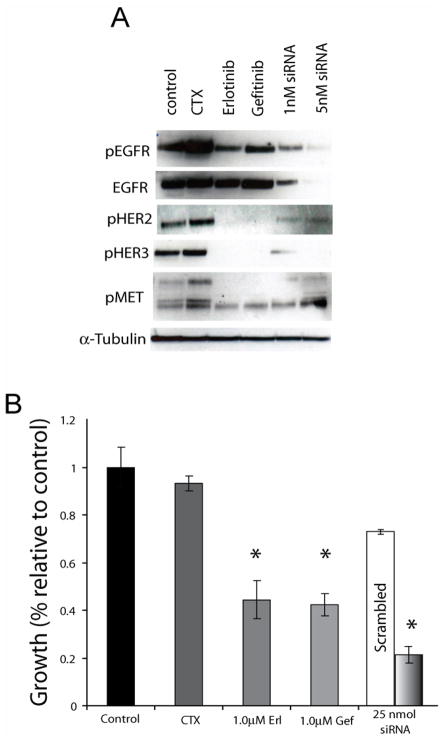
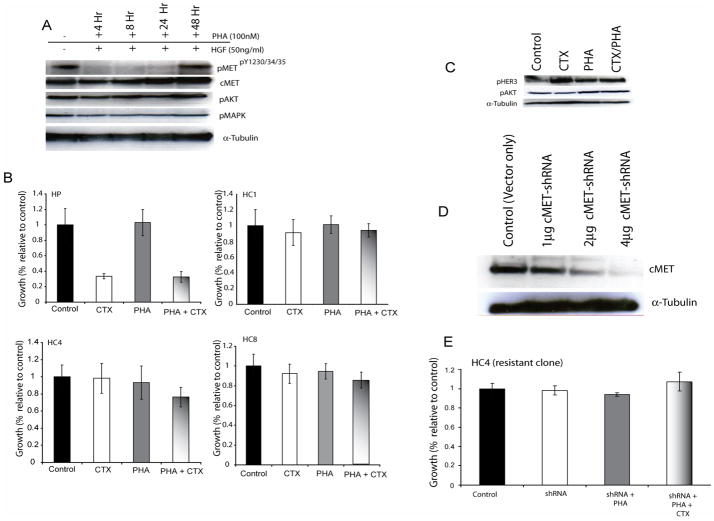
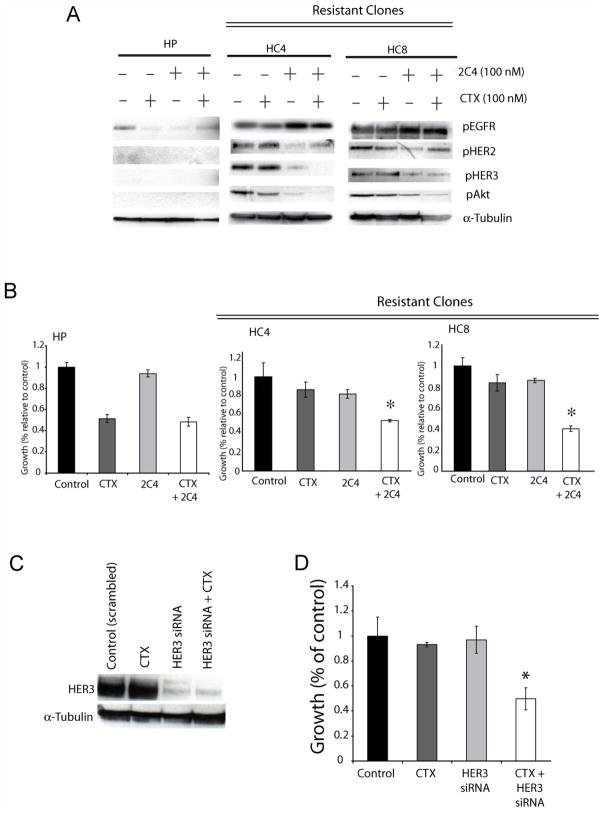
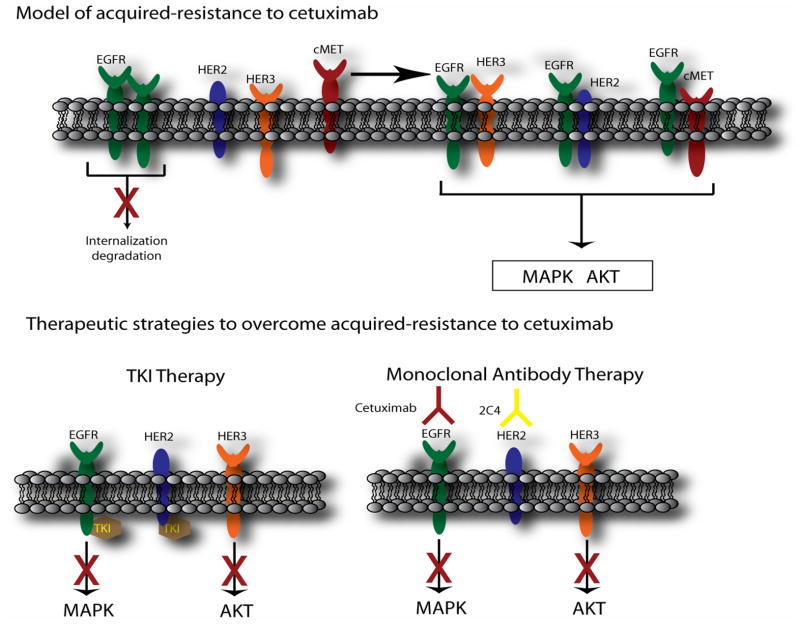
The epidermal growth factor receptor (EGFR) is a central regulator of proliferation and progression in human cancers. Five EGFR inhibitors, two monoclonal antibodies and three TKIs, have recently gained FDA approval in oncology (cetuximab, panitumumab, erlotinib, gefitinib and lapatinib). These strategies of EGFR inhibition demonstrate major tumor regressions in approximately 10-20% of advanced cancer patients. However, many tumors eventually manifest acquired resistance to treatment. In this study we established and characterized a model to study molecular mechanisms of acquired resistance to the EGFR monoclonal antibody cetuximab. Using high-throughput screening we examined the activity of 42 receptor tyrosine kinases in resistant tumor cells following chronic exposure to cetuximab. Cells developing acquired resistance to cetuximab exhibited increased steady-state EGFR expression secondary to alterations in trafficking and degradation. In addition, cetuximab-resistant cells manifested strong activation of HER2, HER3 and cMET. EGFR upregulation promoted increased dimerization with HER2 and HER3 leading to their transactivation. Blockade of EGFR and HER2 led to loss of HER3 and PI(3)K/Akt activity. These data suggest that acquired resistance to cetuximab is accompanied by dysregulation of EGFR internalization/degradation and subsequent EGFR-dependent activation of HER3. Taken together these findings suggest a rationale for the clinical evaluation of combinatorial anti-HER targeting approaches in tumors manifesting acquired resistance to cetuximab.
Figures
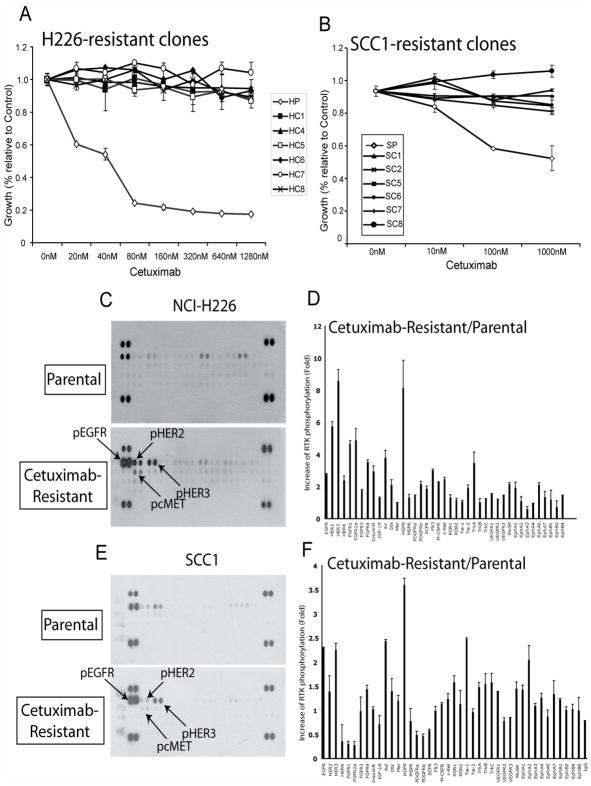
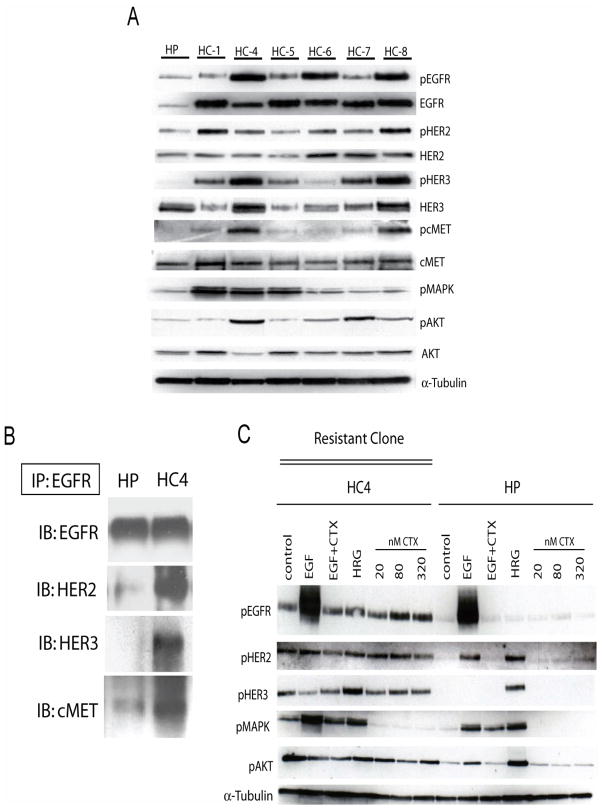
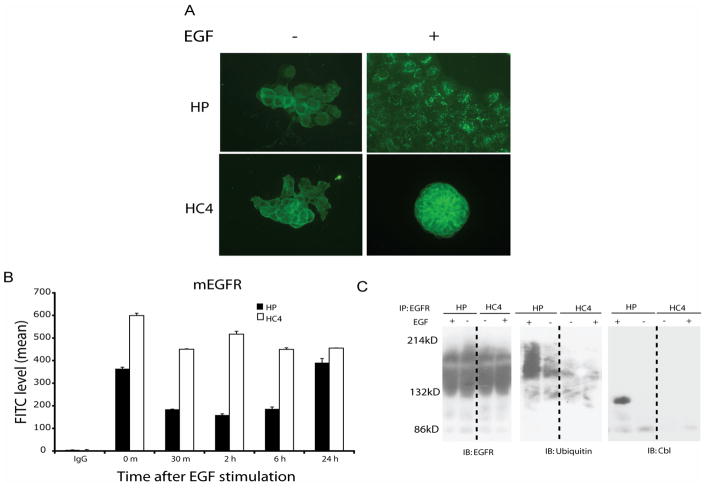
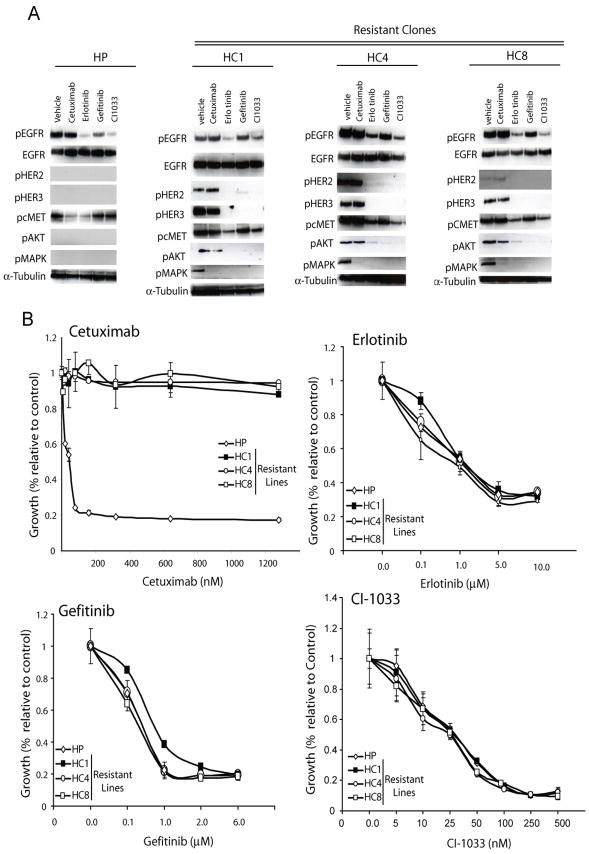
References
-
- Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–59. - PubMed
-
- Benavente S, Huang S, Armstrong E, Chi A, Chinnaiyan P, Harari PM. Establishment of acquired resistance to epidermal growth factor receptor (EGFR) inhibitors in human tumor cell lines. AACR Meeting Abstracts. 2004:1230-a.
-
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. - PubMed
-
- Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63:7345–55. - PubMed
-
- Ciardiello F, Bianco R, Caputo R, Caputo R, Damiano V, Troiani T, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res. 2004;10:784–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
