

Unusual phenotypic features in a patient with a novel splice mutation in the GHRHR gene
- PMID: 18297129
- PMCID: PMC2249753
- DOI: 10.2119/2007-00128.Hilal
Unusual phenotypic features in a patient with a novel splice mutation in the GHRHR gene
Abstract
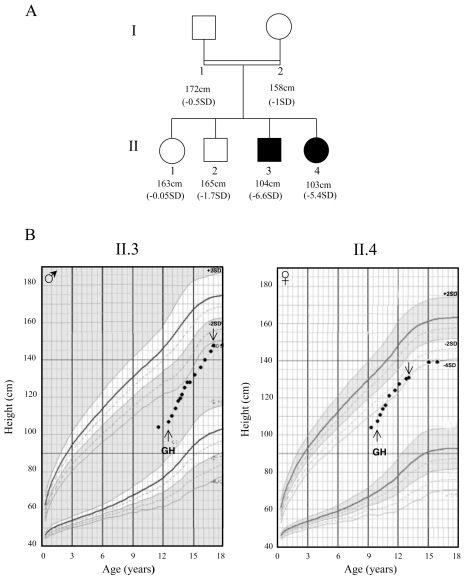
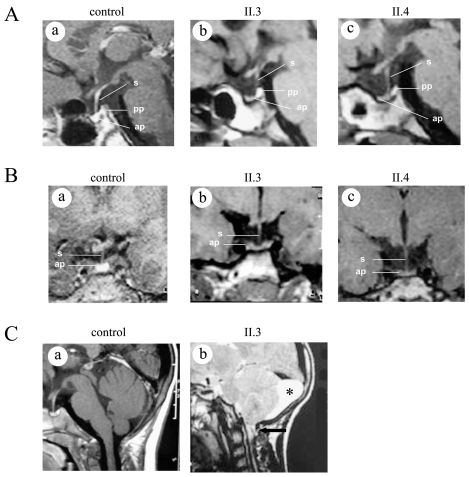
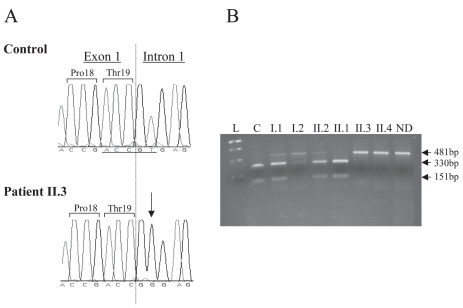
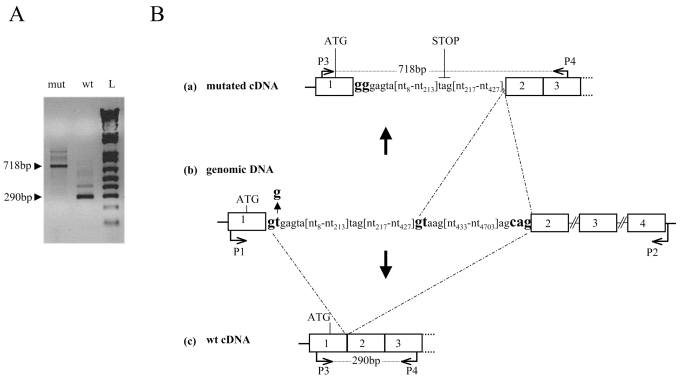
Isolated growth hormone deficiency (IGHD) may be of genetic origin. One of the few genes involved in that condition encodes the growth hormone releasing hormone receptor (GHRHR) that, through its ligand (GHRH), plays a pivotal role in the GH synthesis and secretion by the pituitary. Our objective is to describe the phenotype of two siblings born to a consanguineous union presenting with short stature (IGHD) and Magnetic Resonance Imaging (MRI) abnormalities, and to identify the molecular basis of this condition. Our main outcome measures were clinical and endocrinological investigations, MRI of the pituitary region, study of the GHRHR gene sequence and transcripts. In both patients, the severe growth retardation (-5SD) was combined with anterior pituitary hypoplasia. In addition to these classical phenotypic features for IGHD, one of the patients had a Chiari I malformation, an arachnoid cyst, and a dysmorphic anterior pituitary. A homozygous sequence variation in the consensus donor splice site of intron 1 (IVS1 + 2T > G) of the GHRHR gene was identified in both patients. Using in vitro transcription assay, we showed that this mutation results in abnormal splicing of GHRHR transcripts. In this report, which broadens the phenotype associated with GHRHR defects, we discuss the possible role of the GHRHR in the proper development of extrapituitary structures, through a mechanism that could be direct or secondary to severe GH deficiency.
Figures
References
-
- Alba M, Salvatori R. Familial growth hormone deficiency and mutations in the GHRH receptor gene. Vitam Horm. 2004;69:209–20. - PubMed
-
- Bona G, Paracchini R, Giordano M, Momigliano-Richiardi P. Genetic defects in GH synthesis and secretion. Eur J Endocrinol. 2004;151:S3–9. - PubMed
-
- Cohen RN, et al. Enhanced repression by HESX1 as a cause of hypopituitarism and septooptic dysplasia. J Clin Endocrinol Metab. 2003;88:4832–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
