

Protein abundance profiling of the Escherichia coli cytosol
- PMID: 18304323
- PMCID: PMC2292177
- DOI: 10.1186/1471-2164-9-102
Protein abundance profiling of the Escherichia coli cytosol
Abstract
Background: Knowledge about the abundance of molecular components is an important prerequisite for building quantitative predictive models of cellular behavior. Proteins are central components of these models, since they carry out most of the fundamental processes in the cell. Thus far, protein concentrations have been difficult to measure on a large scale, but proteomic technologies have now advanced to a stage where this information becomes readily accessible.
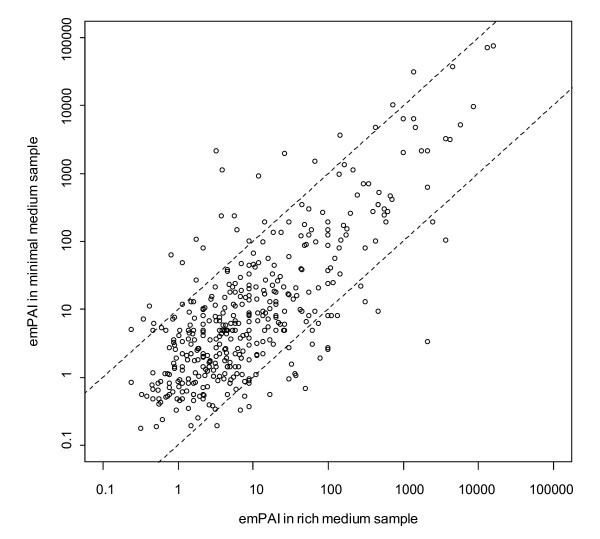
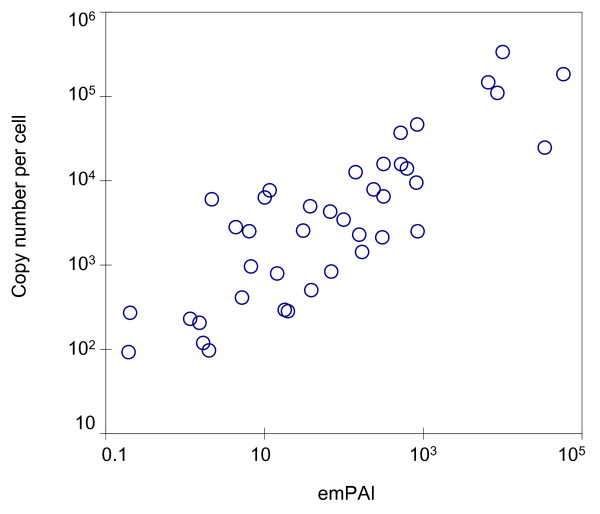
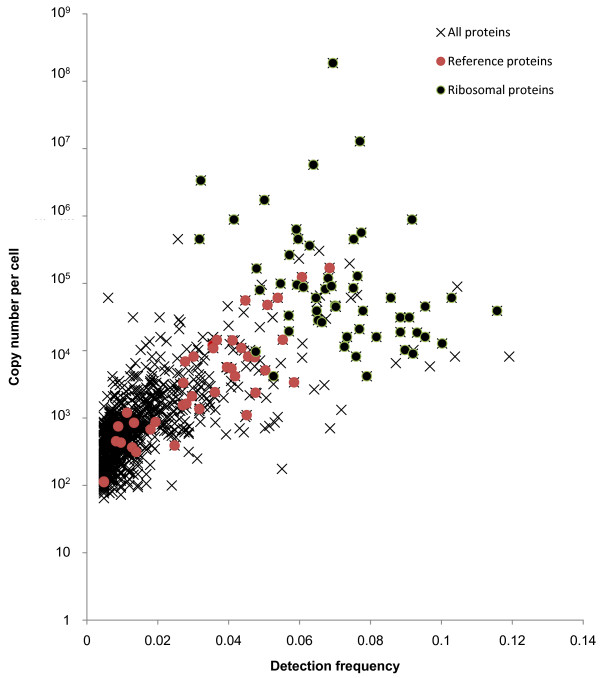
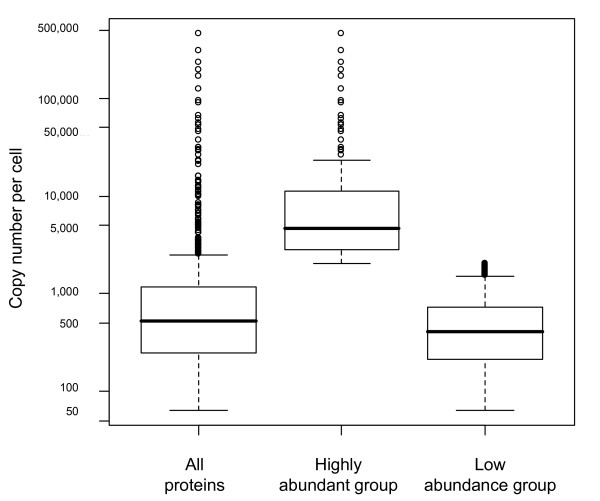
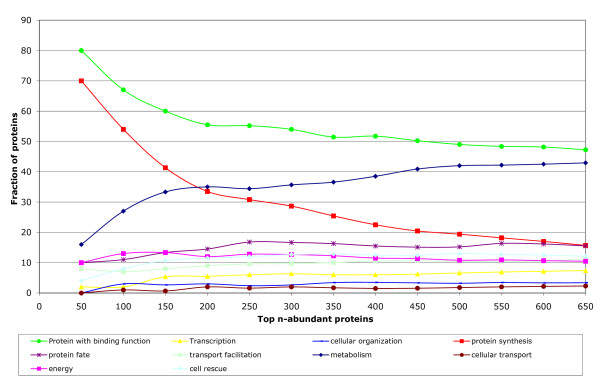
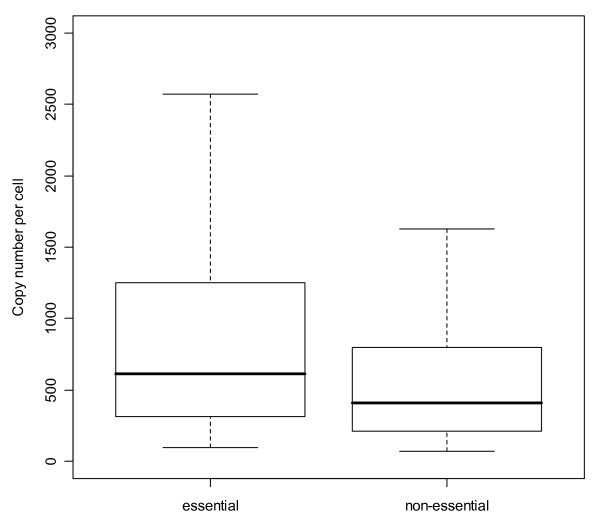
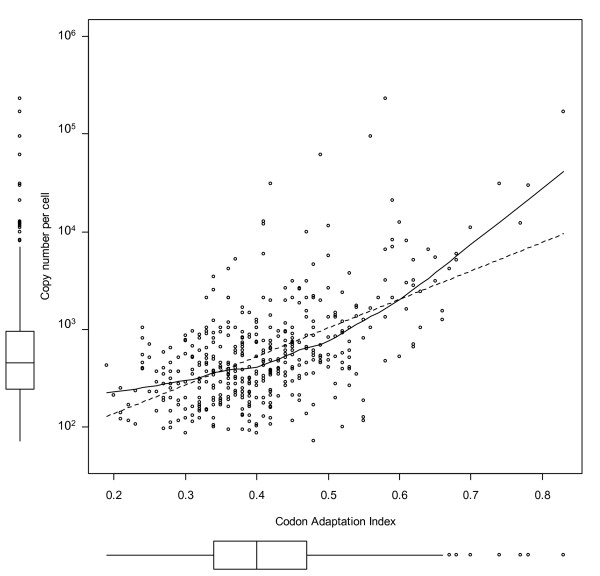
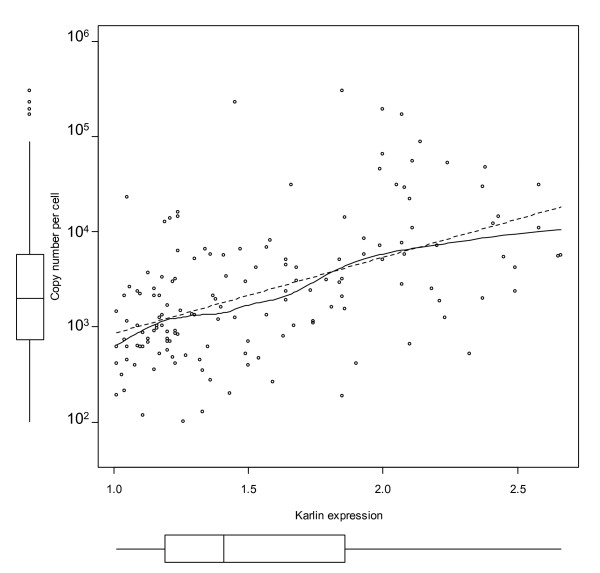
Results: Here, we describe an experimental scheme to maximize the coverage of proteins identified by mass spectrometry of a complex biological sample. Using a combination of LC-MS/MS approaches with protein and peptide fractionation steps we identified 1103 proteins from the cytosolic fraction of the Escherichia coli strain MC4100. A measure of abundance is presented for each of the identified proteins, based on the recently developed emPAI approach which takes into account the number of sequenced peptides per protein. The values of abundance are within a broad range and accurately reflect independently measured copy numbers per cell. As expected, the most abundant proteins were those involved in protein synthesis, most notably ribosomal proteins. Proteins involved in energy metabolism as well as those with binding function were also found in high copy number while proteins annotated with the terms metabolism, transcription, transport, and cellular organization were rare. The barrel-sandwich fold was found to be the structural fold with the highest abundance. Highly abundant proteins are predicted to be less prone to aggregation based on their length, pI values, and occurrence patterns of hydrophobic stretches. We also find that abundant proteins tend to be predominantly essential. Additionally we observe a significant correlation between protein and mRNA abundance in E. coli cells.
Conclusion: Abundance measurements for more than 1000 E. coli proteins presented in this work represent the most complete study of protein abundance in a bacterial cell so far. We show significant associations between the abundance of a protein and its properties and functions in the cell. In this way, we provide both data and novel insights into the role of protein concentration in this model organism.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
