

Sequencing and analysis of the gene-rich space of cowpea
- PMID: 18304330
- PMCID: PMC2279124
- DOI: 10.1186/1471-2164-9-103
Sequencing and analysis of the gene-rich space of cowpea
Abstract
Background: Cowpea, Vigna unguiculata (L.) Walp., is one of the most important food and forage legumes in the semi-arid tropics because of its drought tolerance and ability to grow on poor quality soils. Approximately 80% of cowpea production takes place in the dry savannahs of tropical West and Central Africa, mostly by poor subsistence farmers. Despite its economic and social importance in the developing world, cowpea remains to a large extent an underexploited crop. Among the major goals of cowpea breeding and improvement programs is the stacking of desirable agronomic traits, such as disease and pest resistance and response to abiotic stresses. Implementation of marker-assisted selection and breeding programs is severely limited by a paucity of trait-linked markers and a general lack of information on gene structure and organization. With a nuclear genome size estimated at ~620 Mb, the cowpea genome is an ideal target for reduced representation sequencing.
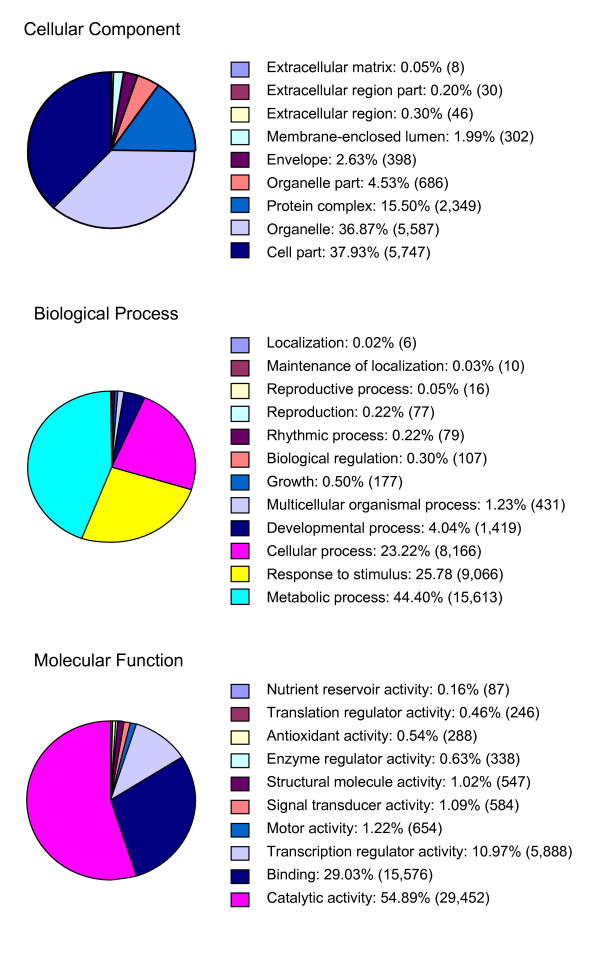
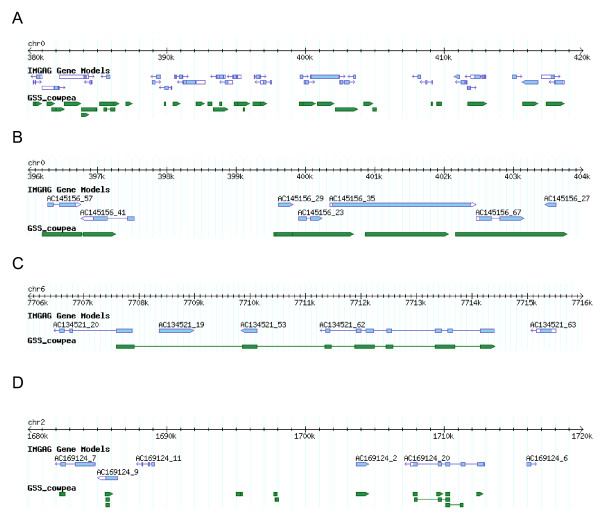
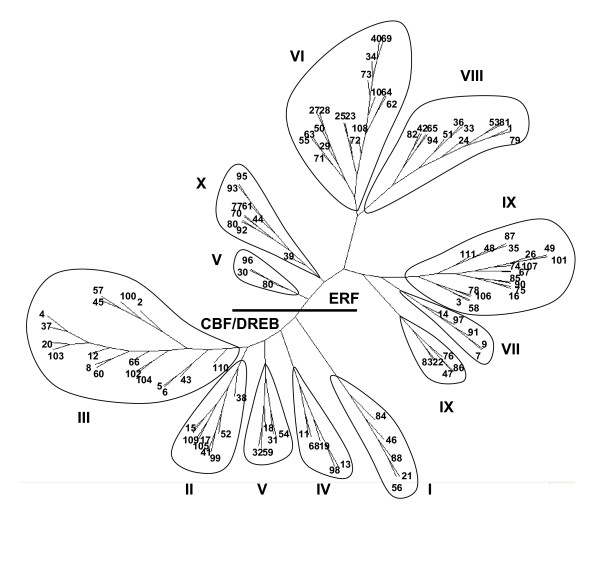
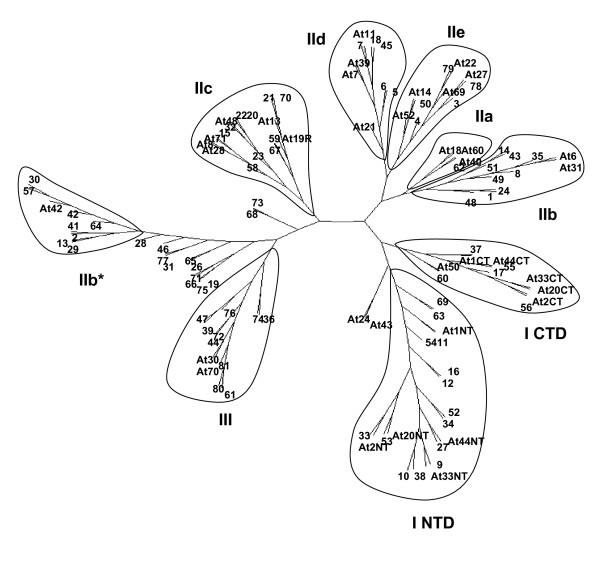
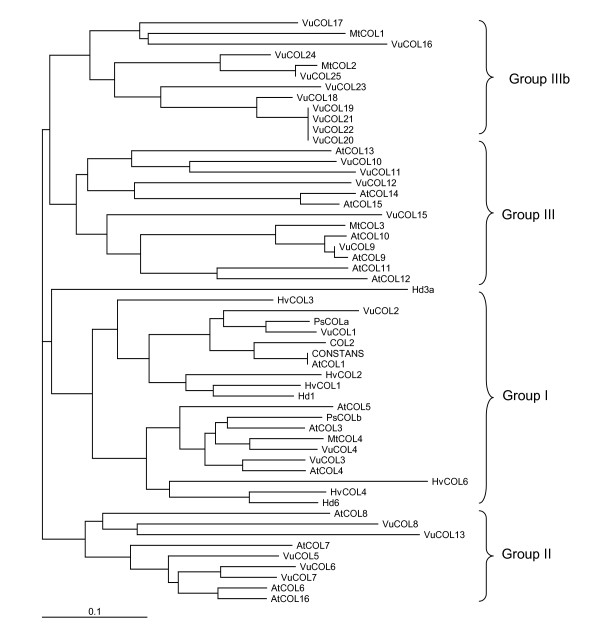
Results: We report here the sequencing and analysis of the gene-rich, hypomethylated portion of the cowpea genome selectively cloned by methylation filtration (MF) technology. Over 250,000 gene-space sequence reads (GSRs) with an average length of 610 bp were generated, yielding ~160 Mb of sequence information. The GSRs were assembled, annotated by BLAST homology searches of four public protein annotation databases and four plant proteomes (A. thaliana, M. truncatula, O. sativa, and P. trichocarpa), and analyzed using various domain and gene modeling tools. A total of 41,260 GSR assemblies and singletons were annotated, of which 19,786 have unique GenBank accession numbers. Within the GSR dataset, 29% of the sequences were annotated using the Arabidopsis Gene Ontology (GO) with the largest categories of assigned function being catalytic activity and metabolic processes, groups that include the majority of cellular enzymes and components of amino acid, carbohydrate and lipid metabolism. A total of 5,888 GSRs had homology to genes encoding transcription factors (TFs) and transcription associated factors (TAFs) representing about 5% of the total annotated sequences in the dataset. Sixty-two (62) of the 64 well-characterized plant transcription factor (TF) gene families are represented in the cowpea GSRs, and these families are of similar size and phylogenetic organization to those characterized in other plants. The cowpea GSRs also provides a rich source of genes involved in photoperiodic control, symbiosis, and defense-related responses. Comparisons to available databases revealed that about 74% of cowpea ESTs and 70% of all legume ESTs were represented in the GSR dataset. As approximately 12% of all GSRs contain an identifiable simple-sequence repeat, the dataset is a powerful resource for the design of microsatellite markers.
Conclusion: The availability of extensive publicly available genomic data for cowpea, a non-model legume with significant importance in the developing world, represents a significant step forward in legume research. Not only does the gene space sequence enable the detailed analysis of gene structure, gene family organization and phylogenetic relationships within cowpea, but it also facilitates the characterization of syntenic relationships with other cultivated and model legumes, and will contribute to determining patterns of chromosomal evolution in the Leguminosae. The micro and macrosyntenic relationships detected between cowpea and other cultivated and model legumes should simplify the identification of informative markers for marker-assisted trait selection and map-based gene isolation necessary for cowpea improvement.
Figures
Similar articles
-
CGKB: an annotation knowledge base for cowpea (Vigna unguiculata L.) methylation filtered genomic genespace sequences.BMC Bioinformatics. 2007 Apr 19;8:129. doi: 10.1186/1471-2105-8-129. BMC Bioinformatics. 2007. PMID: 17445272 Free PMC article.
-
A compendium of transcription factor and Transcriptionally active protein coding gene families in cowpea (Vigna unguiculata L.).BMC Genomics. 2017 Nov 22;18(1):898. doi: 10.1186/s12864-017-4306-1. BMC Genomics. 2017. PMID: 29166879 Free PMC article.
-
The first set of EST resource for gene discovery and marker development in pigeonpea (Cajanus cajan L.).BMC Plant Biol. 2010 Mar 11;10:45. doi: 10.1186/1471-2229-10-45. BMC Plant Biol. 2010. PMID: 20222972 Free PMC article.
-
Molecular genetics of race-specific resistance of cowpea to Striga gesnerioides (Willd.).Pest Manag Sci. 2009 May;65(5):520-7. doi: 10.1002/ps.1722. Pest Manag Sci. 2009. PMID: 19222045 Review.
-
Introgression Breeding in Cowpea [Vigna unguiculata (L.) Walp.].Front Plant Sci. 2020 Sep 16;11:567425. doi: 10.3389/fpls.2020.567425. eCollection 2020. Front Plant Sci. 2020. PMID: 33072144 Free PMC article. Review.
Cited by
-
Recent developments in the chemistry of deoxyribonucleic acid (DNA) intercalators: principles, design, synthesis, applications and trends.Molecules. 2009 May 7;14(5):1725-46. doi: 10.3390/molecules14051725. Molecules. 2009. PMID: 19471193 Free PMC article. Review.
-
Genetic Studies on the Inheritance of Storage-Induced Cooking Time in Cowpeas [Vigna unguiculata (L.) Walp].Front Plant Sci. 2020 May 5;11:444. doi: 10.3389/fpls.2020.00444. eCollection 2020. Front Plant Sci. 2020. PMID: 32431718 Free PMC article.
-
Post-genomics studies of developmental processes in legume seeds.Plant Physiol. 2009 Nov;151(3):1023-9. doi: 10.1104/pp.109.143966. Epub 2009 Aug 12. Plant Physiol. 2009. PMID: 19675147 Free PMC article. Review. No abstract available.
-
SNP discovery in swine by reduced representation and high throughput pyrosequencing.BMC Genet. 2008 Dec 4;9:81. doi: 10.1186/1471-2156-9-81. BMC Genet. 2008. PMID: 19055830 Free PMC article.
-
The genome assembly of asparagus bean, Vigna unguiculata ssp. sesquipedialis.Sci Data. 2019 Jul 17;6(1):124. doi: 10.1038/s41597-019-0130-6. Sci Data. 2019. PMID: 31316072 Free PMC article.
References
-
- Singh BB. Cowpea Vigna unguiculata (L.) Walp. In: Singh RJ, Jauhar PP, editor. Genetic Resources, Chromosome Engineering and Crop Improvement. Vol. 1. Boca Raton: CRC Press; 2005. pp. 117–162.
-
- Timko MP, Ehlers JD, Roberts PA. Cowpea. In: Kole C, editor. Genome Mapping and Molecular Breeding in Plants, Pulses, Sugar and Tuber Crops. Vol. 3. Berlin: Springer-Verlag; 2007. pp. 49–68.
-
- Phillips RD, McWatters KH, Chinnan J, Komey NS, Liu K, Mensa-Wilmot Y, Nnanna IA, Okeke C, Prinyawiwatkul W, Saalia FK. Utilization of cowpea for human food. Field Crops Res. 2003;82:193–213.
-
- Lewis G, Schire B, Mackinder B, Lock M. Legumes of the World. London: Kew Publishing; 2005.
-
- Lavin M, Herendeen PS, Wojciechowski MF. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst Biol. 2005;54:575–594. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
