PARTS: probabilistic alignment for RNA joinT secondary structure prediction
- PMID: 18304945
- PMCID: PMC2367733
- DOI: 10.1093/nar/gkn043
PARTS: probabilistic alignment for RNA joinT secondary structure prediction
Abstract
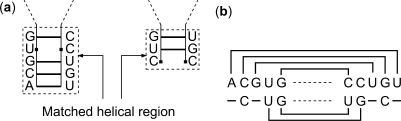
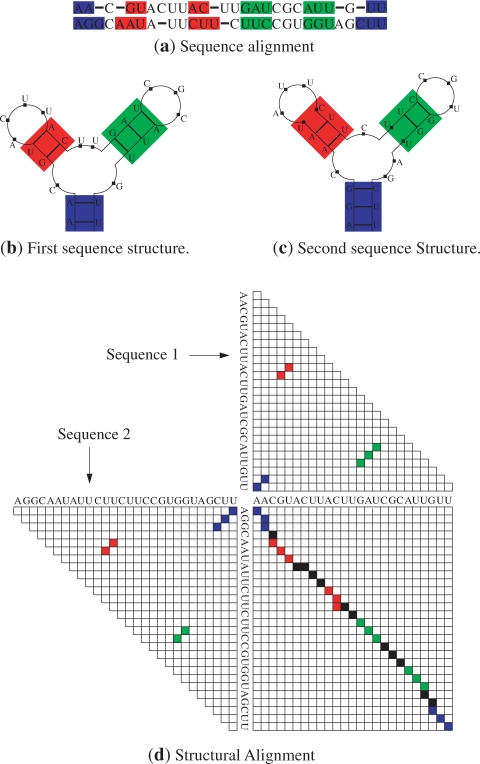
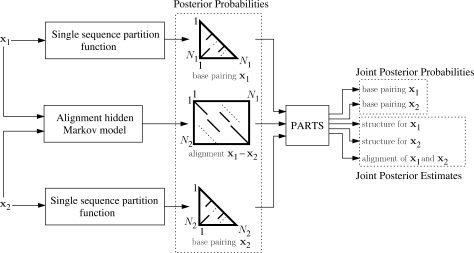
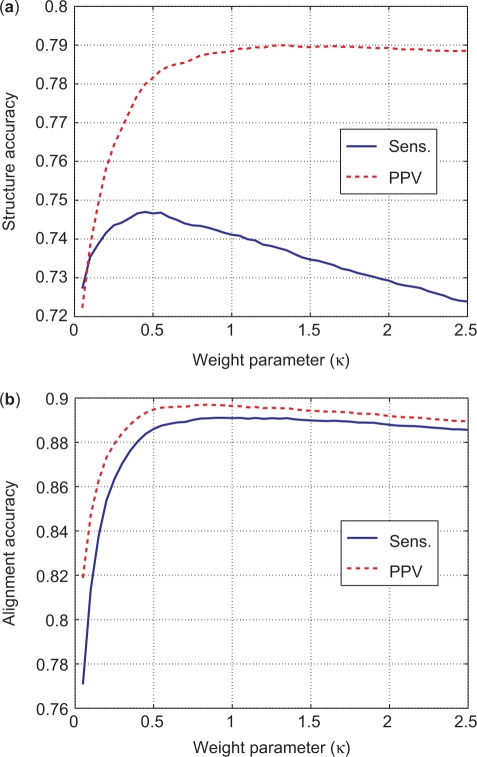
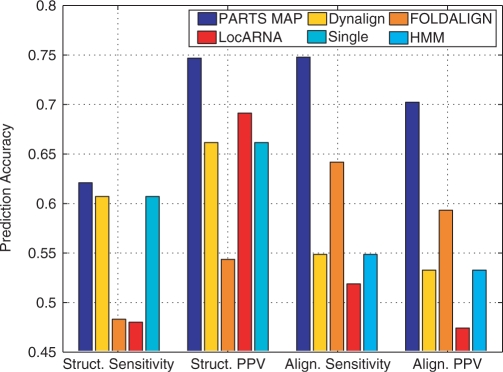
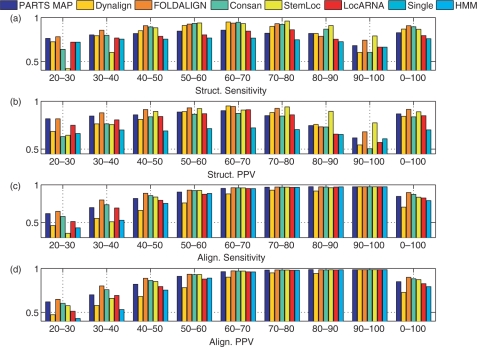
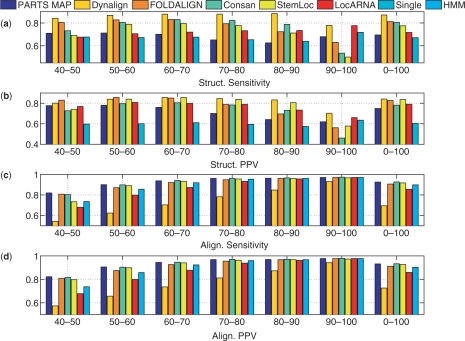
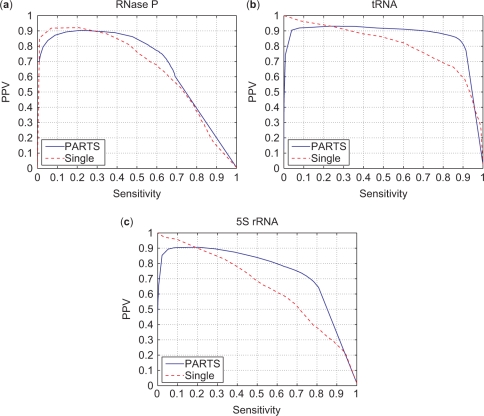
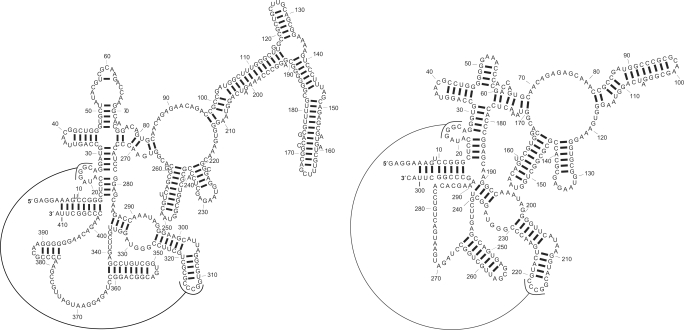
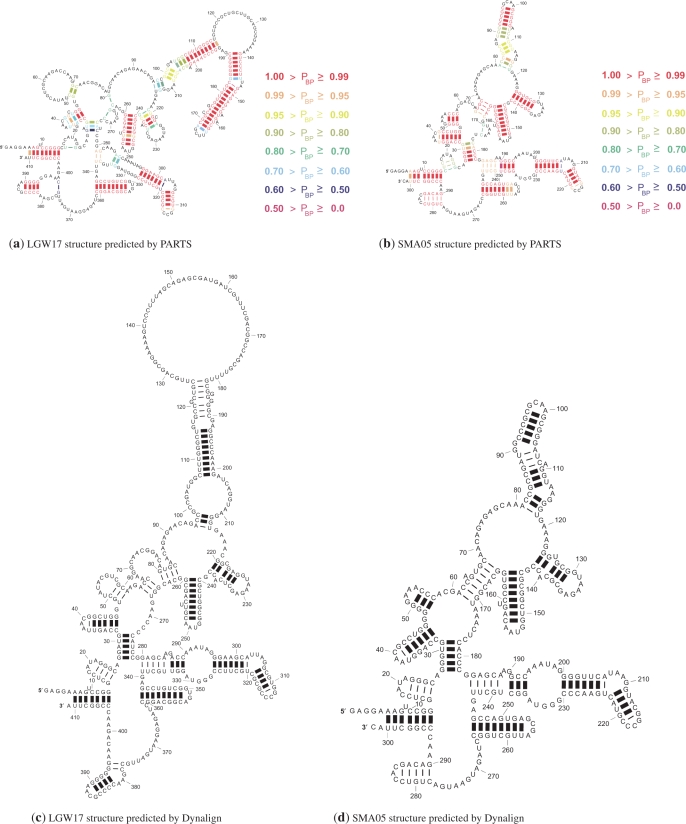
A novel method is presented for joint prediction of alignment and common secondary structures of two RNA sequences. The joint consideration of common secondary structures and alignment is accomplished by structural alignment over a search space defined by the newly introduced motif called matched helical regions. The matched helical region formulation generalizes previously employed constraints for structural alignment and thereby better accommodates the structural variability within RNA families. A probabilistic model based on pseudo free energies obtained from precomputed base pairing and alignment probabilities is utilized for scoring structural alignments. Maximum a posteriori (MAP) common secondary structures, sequence alignment and joint posterior probabilities of base pairing are obtained from the model via a dynamic programming algorithm called PARTS. The advantage of the more general structural alignment of PARTS is seen in secondary structure predictions for the RNase P family. For this family, the PARTS MAP predictions of secondary structures and alignment perform significantly better than prior methods that utilize a more restrictive structural alignment model. For the tRNA and 5S rRNA families, the richer structural alignment model of PARTS does not offer a benefit and the method therefore performs comparably with existing alternatives. For all RNA families studied, the posterior probability estimates obtained from PARTS offer an improvement over posterior probability estimates from a single sequence prediction. When considering the base pairings predicted over a threshold value of confidence, the combination of sensitivity and positive predictive value is superior for PARTS than for the single sequence prediction. PARTS source code is available for download under the GNU public license at http://rna.urmc.rochester.edu.
Figures