

Review
doi: 10.1016/j.neuropharm.2008.01.005.
Epub 2008 Jan 25.
Mechanisms of ischemic brain damage
Affiliations
- PMID: 18308346
- PMCID: PMC2603601
- DOI: 10.1016/j.neuropharm.2008.01.005
Item in Clipboard
Review
Mechanisms of ischemic brain damage
Neuropharmacology.
2008 Sep.
Abstract
In the United States stroke is the third leading cause of death and the leading cause of disability. Brain injury following stroke results from the complex interplay of multiple pathways including excitotoxicity, acidotoxicity, ionic imbalance, peri-infarct depolarization, oxidative and nitrative stress, inflammation and apoptosis. There are very few treatments for stroke and the development of new treatments requires a comprehensive understanding of the diverse mechanisms of ischemic brain damage that are responsible for neuronal death. Here, we discuss the underlying pathophysiology of this devastating disease and reveal the intertwined pathways that are the target of therapeutic intervention.
Figures
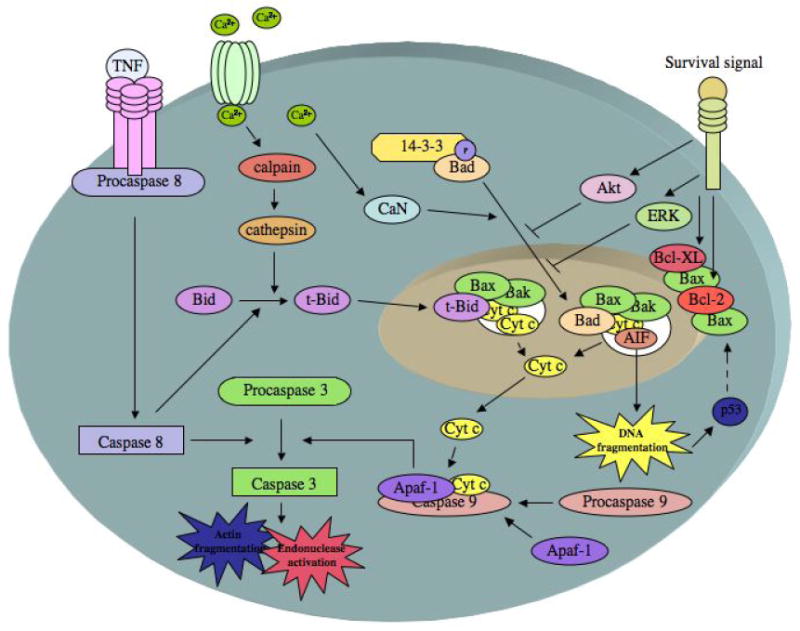
Cell death pathways relevant to apoptosis in cerebral ischemia. The release of cytochrome c (cyt c) from the mitochondria is mediated by the proapoptotic proteins Bax and/or Bak forming a pore in the mitochondrial membrane. Pore formation is facilitated by Bad and Bid. Calcium influx causes the dephosphorylation of Bad by calcineurin (CaN), which releases BAD from 14-3-3 and allows its translocation to the mitochondria. Calcium can also activate calpains, which can activate cathepsins that mediate the limited proteolysis of Bid, allowing truncated Bid (t-Bid) to translocate to the mitochondria. Caspase 8, which is activated by TNF receptor ligation, also mediates the limited proteolysis of Bid, allowing truncated Bid (t-Bid) to translocate to the mitochondria. Once present in the cytosol, cyt c forms the apoptosome complex by binding to Apaf-1 and procaspase 9. The apoptosome complex cleaves and activates caspase 3, which causes actin fragmentation, and endonuclease activation. Caspase 3 can also be activated by caspase 8. Apoptosis-inducing factor (AIF) can also be released from the pore created in the mitochondria, causing DNA degradation. DNA damage activates p53, which further increases Bax expression. The antiapoptotic proteins Bcl-XL and Bcl-2 prevent Bax mediated pore formation and cyt c release. Various survival factors also prevent pore formation and cytochrome C release by activation of Akt and ERK pathways (adapted from (Edvinsson and Krause 2002)).
References
-
- Adams J, Cory S. Life or death decisions by the Bcl-2 protein family. Trends Biochem Sci. 2001;26:61–66. - PubMed
-
- Antonsson B, Montessuit S, et al. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. Journal of Biological Chemistry. 2001;276:11615–11623. - PubMed
-
- Auer R. Combination therapy with U74006F (tirilazad mesylate), MK-801, insulin and diazepam in transient forebrain ischemia. Neurology Research. 1995;17:132–136. - PubMed
-
- Bordet R, Deplanque D, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab. 2000;20:1190–6. - PubMed
-
- Bruce AJ, Boling W, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nature Medicine. 1996;2:788–794. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
