

Immune surveillance: a balance between protumor and antitumor immunity
- PMID: 18308558
- PMCID: PMC2699403
- DOI: 10.1016/j.gde.2007.12.007
Immune surveillance: a balance between protumor and antitumor immunity
Abstract
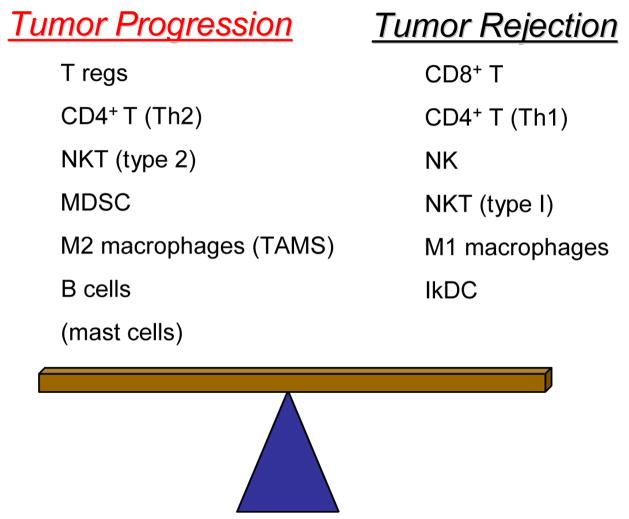
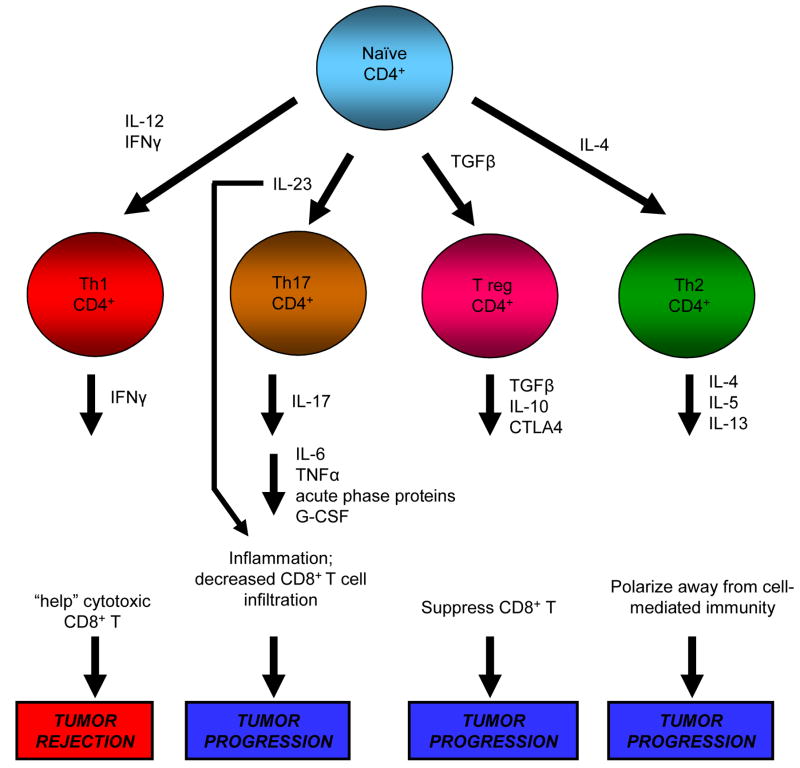
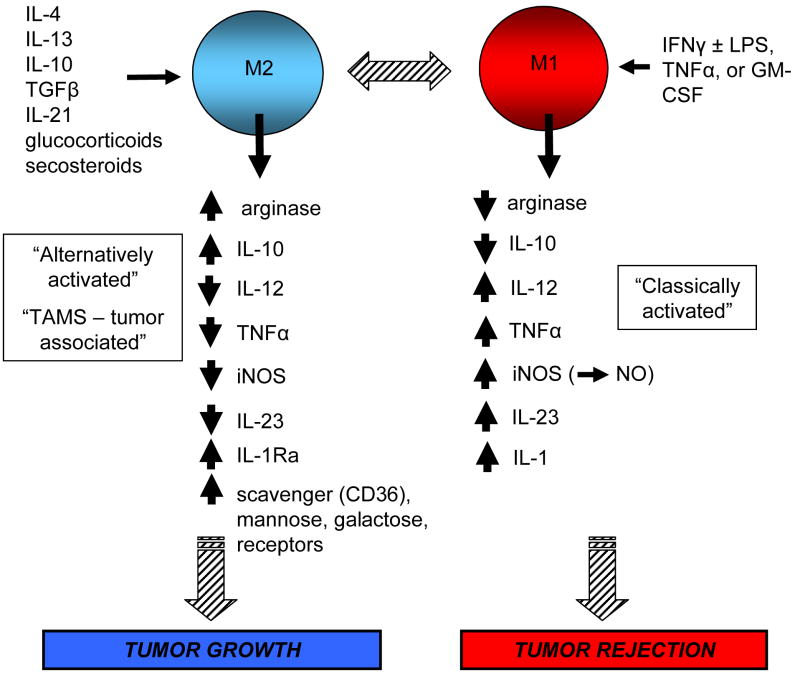
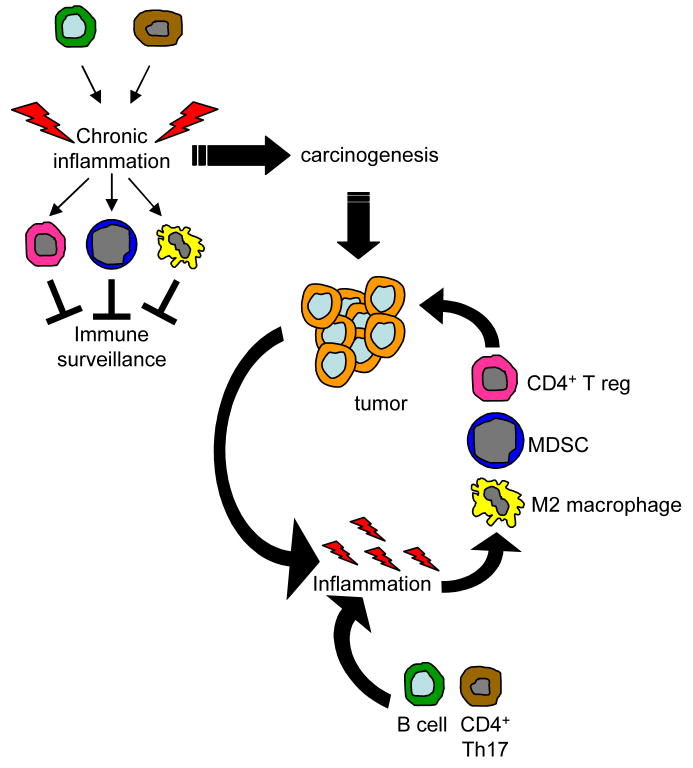
Precancerous and malignant cells can induce an immune response which results in the destruction of transformed and/or malignant cells, a process known as immune surveillance. However, immune surveillance is not always successful, resulting in 'edited' tumors that have escaped immune surveillance. Immunoediting is not simply because of the absence of antitumor immunity, but is because of protumor immunity that blocks antitumor adaptive and innate responses, and promotes conditions that favor tumor progression. Several immune protumor effector mechanisms are upregulated by chronic inflammation, leading to the hypothesis that inflammation promotes carcinogenesis and tumor growth by altering the balance between protumor and antitumor immunity, thereby preventing the immune system from rejecting malignant cells, and providing a tumor-friendly environment for progressive disease.
Figures
References
-
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. - PubMed
-
- Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. This is a comprehensive review of the concepts of cancer immunosurveillance and immunoediting by the investigators who resurrected the immunosurveillance hypothesis. - PubMed
-
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
